Annotation-Gap Discovery via Phenotype-Fitness-Pangenome-Gapfilling Integration
CompletedResearch Question
Can we systematically identify and resolve metabolic annotation gaps in bacterial genomes by integrating experimental growth phenotypes, gene fitness data, metabolic model gapfilling, pangenome context, and sequence homology?
Research Plan
Hypothesis
- H0: Gapfilled reactions required to reconcile observed growth phenotypes with metabolic model predictions cannot be assigned to specific genes using fitness data, pangenome co-occurrence, and exemplar sequence homology — the annotation gaps are genuine knowledge gaps with no tractable candidates.
- H1: A significant fraction of gapfilled reactions can be confidently assigned to specific genes by triangulating (A) BLAST/DIAMOND homology to known exemplar sequences, (B) gene fitness evidence under the relevant carbon source, and (C) pangenome co-occurrence with known pathway genes — resolving annotation gaps and improving model-phenotype agreement.
Approach
Overview
- Select ~20 genomes from Fitness Browser organisms with rich carbon-source experiment coverage, cross-referenced with BacDive growth phenotypes
- Build draft metabolic models using ModelSEEDpy from RAST/Bakta annotations
- Simulate growth on each carbon source via FBA (COBRApy) and compare to observed phenotypes
- Conditional gapfilling for false-negative cases (observed growth, predicted no-growth)
- GapMind pathway analysis to identify missing pathway steps independently
- Fitness-guided candidate identification using RB-TnSeq data under carbon-source conditions
- Exemplar BLAST of gapfilled-reaction protein sequences against target proteomes
- Triangulated gene-reaction assignment with confidence scoring
- Validation by re-running FBA with new GPRs and measuring improvement
Data Sources
| Source | Database | What it provides |
|---|---|---|
| Growth phenotypes | kescience_fitnessbrowser.experiment |
Carbon source conditions with fitness data |
| Growth phenotypes | kescience_bacdive.metabolite_utilization |
Binary +/- utilization for 988K strain-compound pairs |
| Gene fitness | kescience_fitnessbrowser.genefitness |
Per-gene fitness scores under carbon source conditions |
| Metabolic models | ModelSEEDpy (installed locally) | Draft GEM construction from genome annotations |
| FBA/Gapfilling | COBRApy (installed locally) | Flux balance analysis and conditional gapfilling |
| Reference biochemistry | kbase_msd_biochemistry |
56K reactions, 46K molecules, stoichiometry |
| Pathway predictions | kbase_ke_pangenome.gapmind_pathways |
Carbon + amino acid pathway completeness per genome |
| Pangenome clusters | kbase_ke_pangenome.gene_cluster |
Gene families, presence/absence, representative sequences |
| Functional annotations | kbase_ke_pangenome.eggnog_mapper_annotations |
EC, KEGG, COG for gene clusters |
| Protein annotations | kbase_ke_pangenome.bakta_annotations |
EC, UniRef, product descriptions |
| Cross-references | kbase_ke_pangenome.bakta_db_xrefs |
UniRef, KEGG, Pfam cross-references (572M rows) |
| Exemplar sequences | UniProt/Swiss-Prot via UniRef mappings | Reviewed protein sequences for gapfilled reactions |
Revision History
- v1 (2026-05-07): Initial plan
Overview
Genome-scale metabolic models built from automated annotation pipelines routinely require gapfilling to match observed growth phenotypes. Each gapfilled reaction represents an "annotation gap" — a metabolic function the organism performs but whose responsible gene remains unidentified. This project integrates five independent evidence types to resolve these gaps:
- ModelSEED/COBRApy models identify which reactions are missing (gapfilling)
- GapMind pathway predictions identify missing pathway steps independently
- Fitness Browser RB-TnSeq data reveals which genes matter under each carbon source
- Pangenome gene co-occurrence links candidates to known pathway genes
- DIAMOND BLAST against exemplar sequences confirms sequence homology
By triangulating these evidence types, we assign confidence-scored gene-reaction links to previously orphan gapfilled reactions, improving metabolic model accuracy and reducing annotation debt. The study covers 14 organisms (selected from 48 Fitness Browser organisms based on carbon-source experiment coverage, taxonomic diversity, and successful model building) across 18 carbon sources.
Key Findings
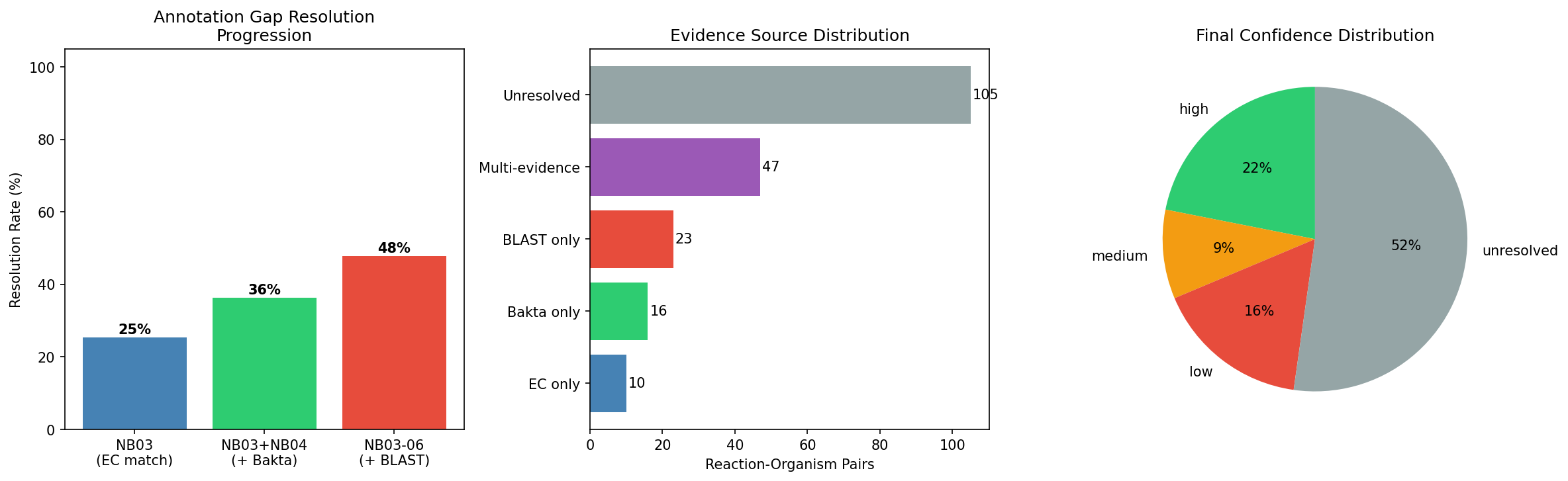
1. Evidence Triangulation Resolves 47.8% of Annotation Gaps
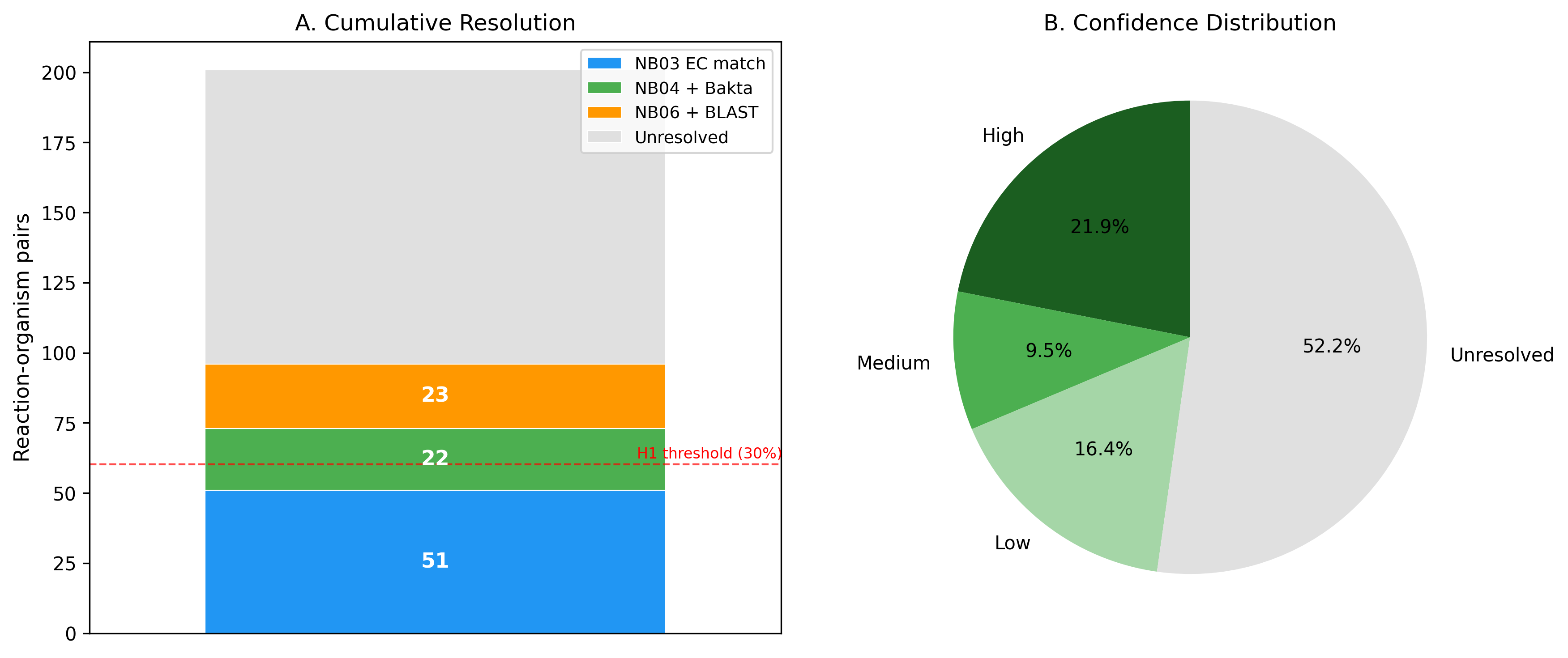
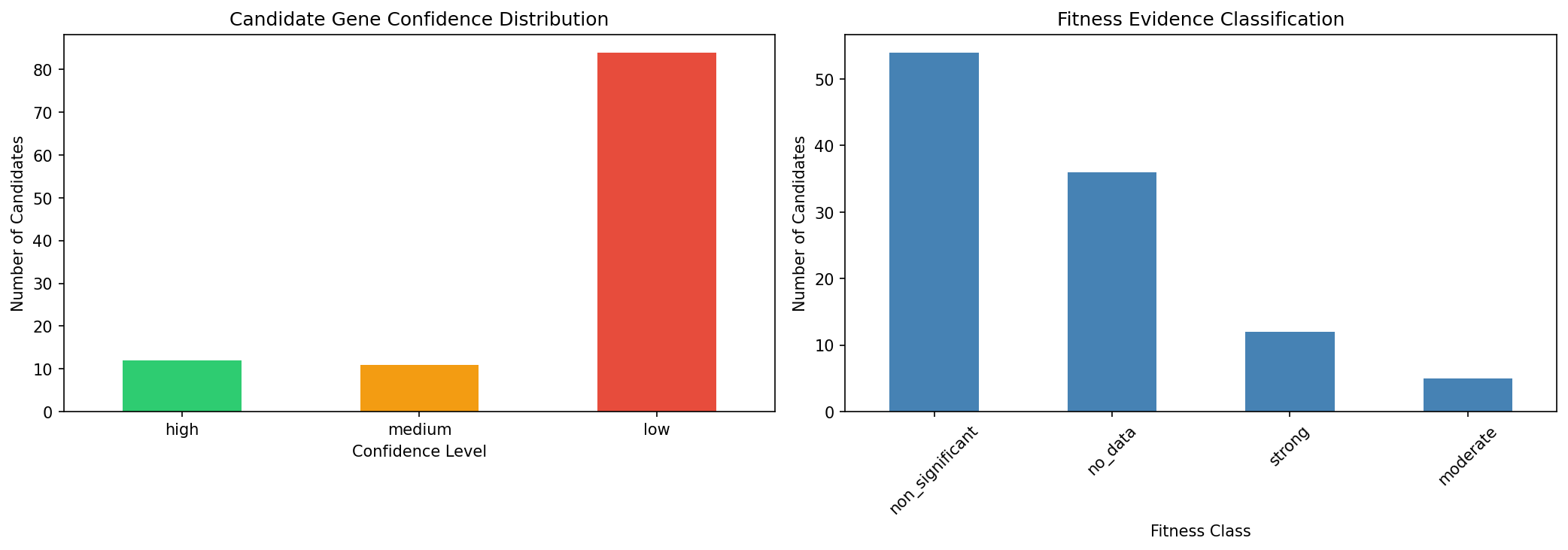
Of 201 gapfilled enzymatic reaction-organism pairs across 14 Fitness Browser organisms and 18 carbon sources, 96 (47.8%) were assigned candidate genes with confidence scoring. This exceeds the pre-specified H1 threshold of 30%, supporting the hypothesis that integrating multiple evidence types can resolve a significant fraction of metabolic annotation gaps.
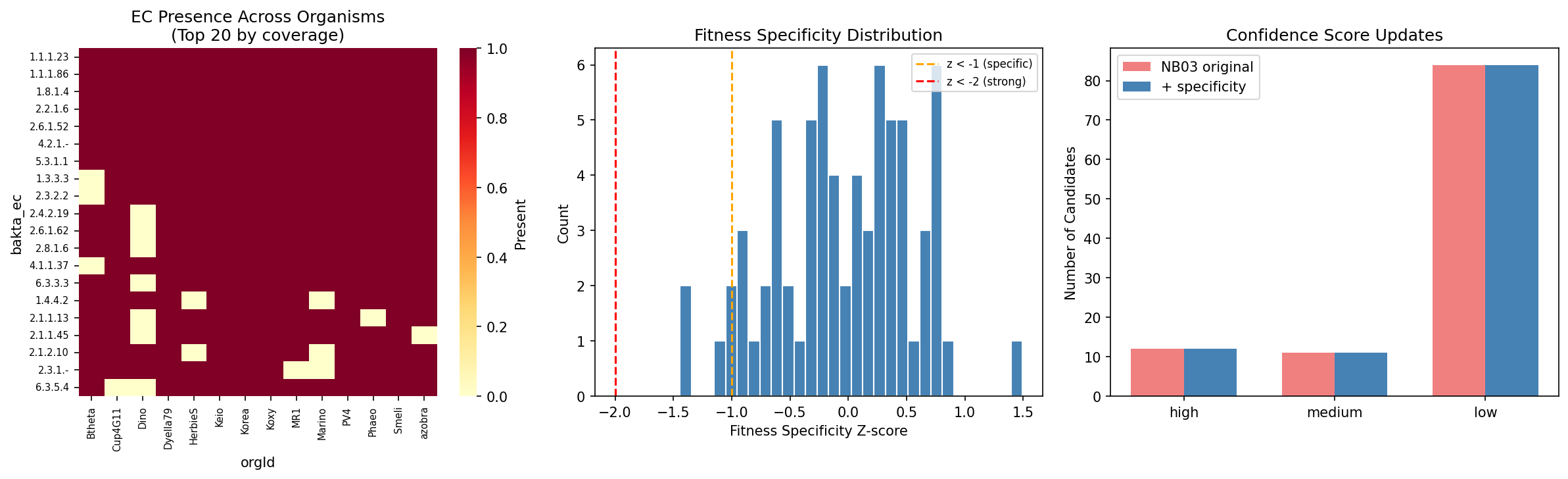
- High confidence: 44 pairs (21.9%) — BLAST homology + fitness evidence + pangenome conservation
- Medium confidence: 19 pairs (9.5%) — BLAST homology with partial supporting evidence
- Low confidence: 33 pairs (16.4%) — single evidence stream only
- Unresolved: 105 pairs (52.2%) — no convincing gene candidate identified
(Notebook: 06_exemplar_blast.ipynb, 07_validation_analysis.ipynb)
2. No Single Evidence Stream Achieves >35% Resolution
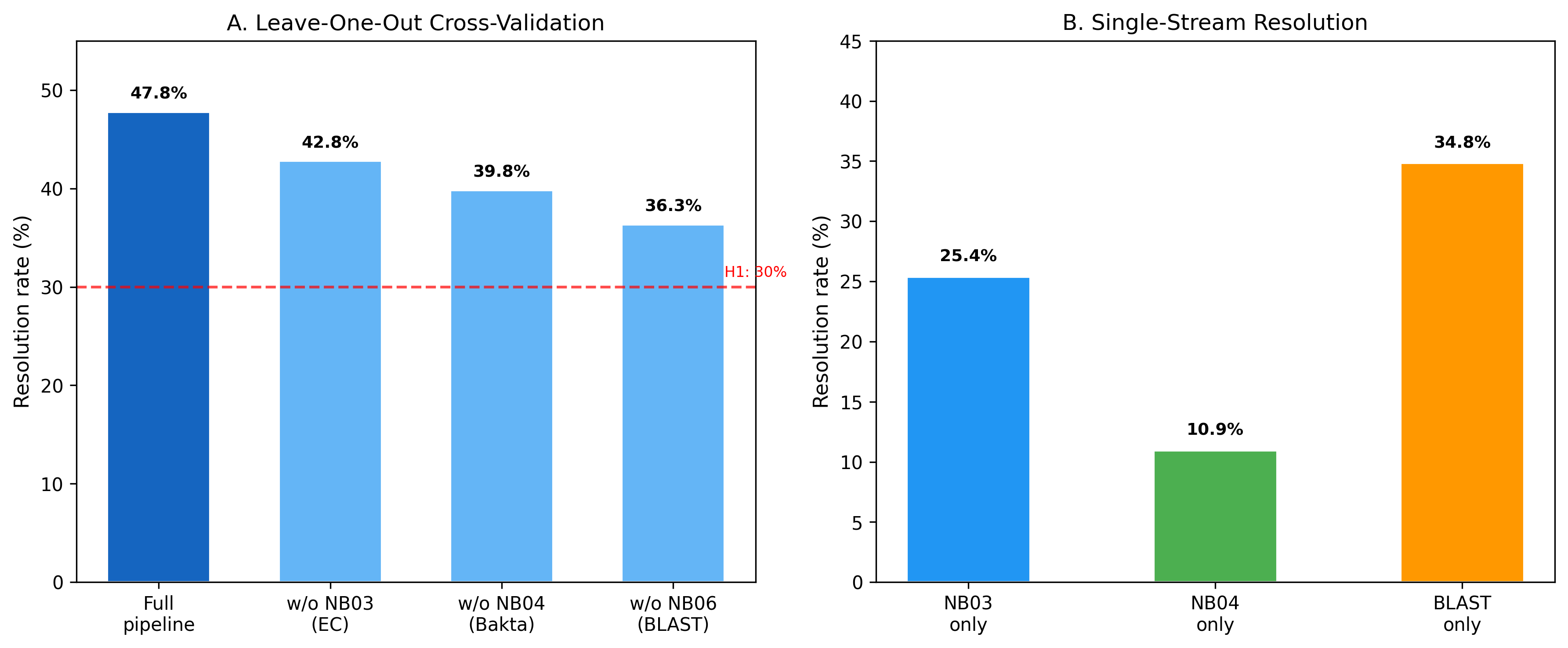
Leave-one-out cross-validation demonstrates that each evidence stream contributes uniquely to resolution:
| Configuration | Resolved | Rate |
|---|---|---|
| Full pipeline | 96 | 47.8% |
| Without NB03 (EC match) | 86 | 42.8% |
| Without NB04 (Bakta) | 80 | 39.8% |
| Without NB06 (BLAST) | 73 | 36.3% |
| NB03 alone | 51 | 25.4% |
| NB04 alone | 22 | 10.9% |
| BLAST alone | 70 | 34.8% |
BLAST homology was the single most impactful stream (34.8% alone), but the full pipeline adds 13 percentage points over BLAST alone. Removing any single stream still keeps resolution above 36%, demonstrating robustness.
(Notebook: 07_validation_analysis.ipynb)
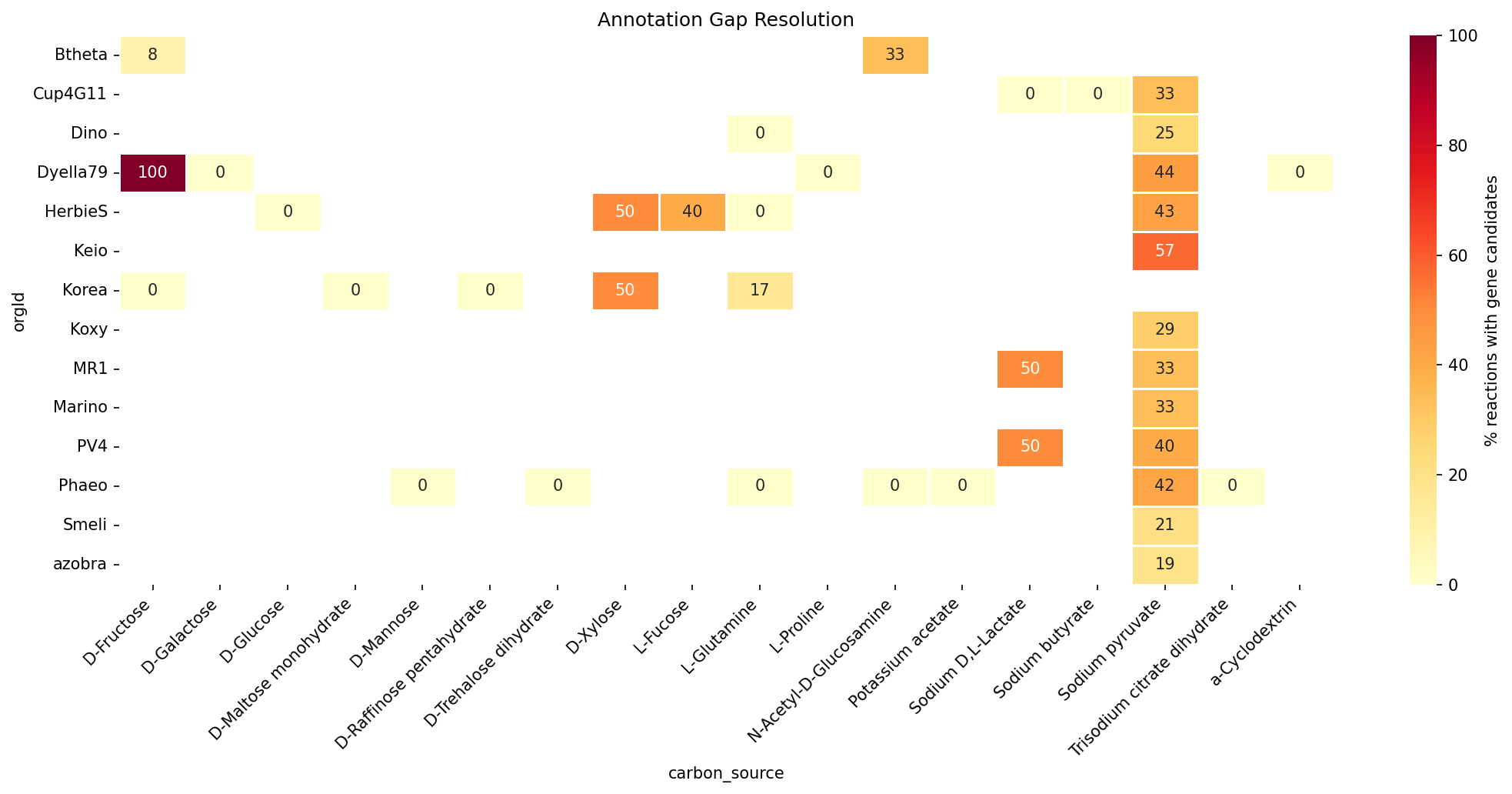
3. Resolution Varies 3.5-fold Across Organisms
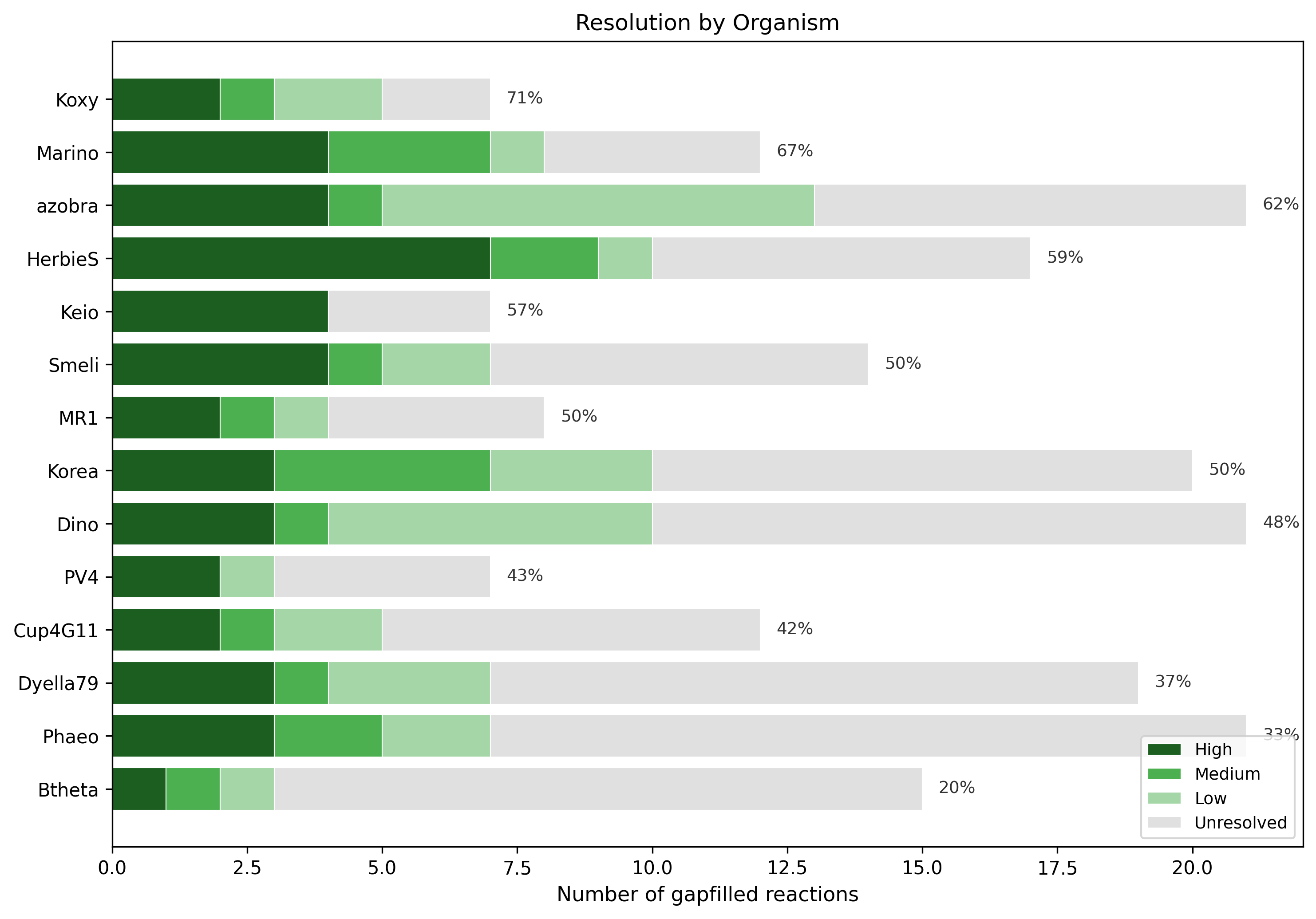
Resolution rates ranged from 20% (Bacteroides thetaiotaomicron) to 71% (Klebsiella michiganensis). Organisms with better-annotated reference genomes and stronger Fitness Browser coverage showed higher resolution rates. B. thetaiotaomicron — a Bacteroidetes with more divergent metabolism — had the lowest resolution, consistent with its phylogenetic distance from the proteobacterial majority.
| Organism | Total Gaps | Resolved | Rate |
|---|---|---|---|
| K. michiganensis (Koxy) | 7 | 5 | 71.4% |
| Marinobacter (Marino) | 12 | 8 | 66.7% |
| Azospirillum brasilense (azobra) | 21 | 13 | 61.9% |
| Herbaspirillum seropedicae (HerbieS) | 17 | 10 | 58.8% |
| E. coli Keio (Keio) | 7 | 4 | 57.1% |
| B. thetaiotaomicron (Btheta) | 15 | 3 | 20.0% |
(Notebook: 08_figures_report.ipynb)
4. Two Reactions Dominate High-Confidence Assignments
The reactions rxn02185 (2-acetolactate pyruvate-lyase, EC 2.2.1.6, branched-chain amino acid biosynthesis) and rxn03436 (acetohydroxy acid isomeroreductase, EC 1.1.1.86, valine/isoleucine biosynthesis) were each resolved with high confidence in 9 of 14 organisms. These enzymes catalyze sequential steps in the same biosynthetic pathway, and their consistent co-resolution validates the triangulation approach — when one pathway gene is found, the adjacent step's gene is typically recoverable.
(Notebook: 06_exemplar_blast.ipynb)
5. "Dark Reactions" Resist Resolution
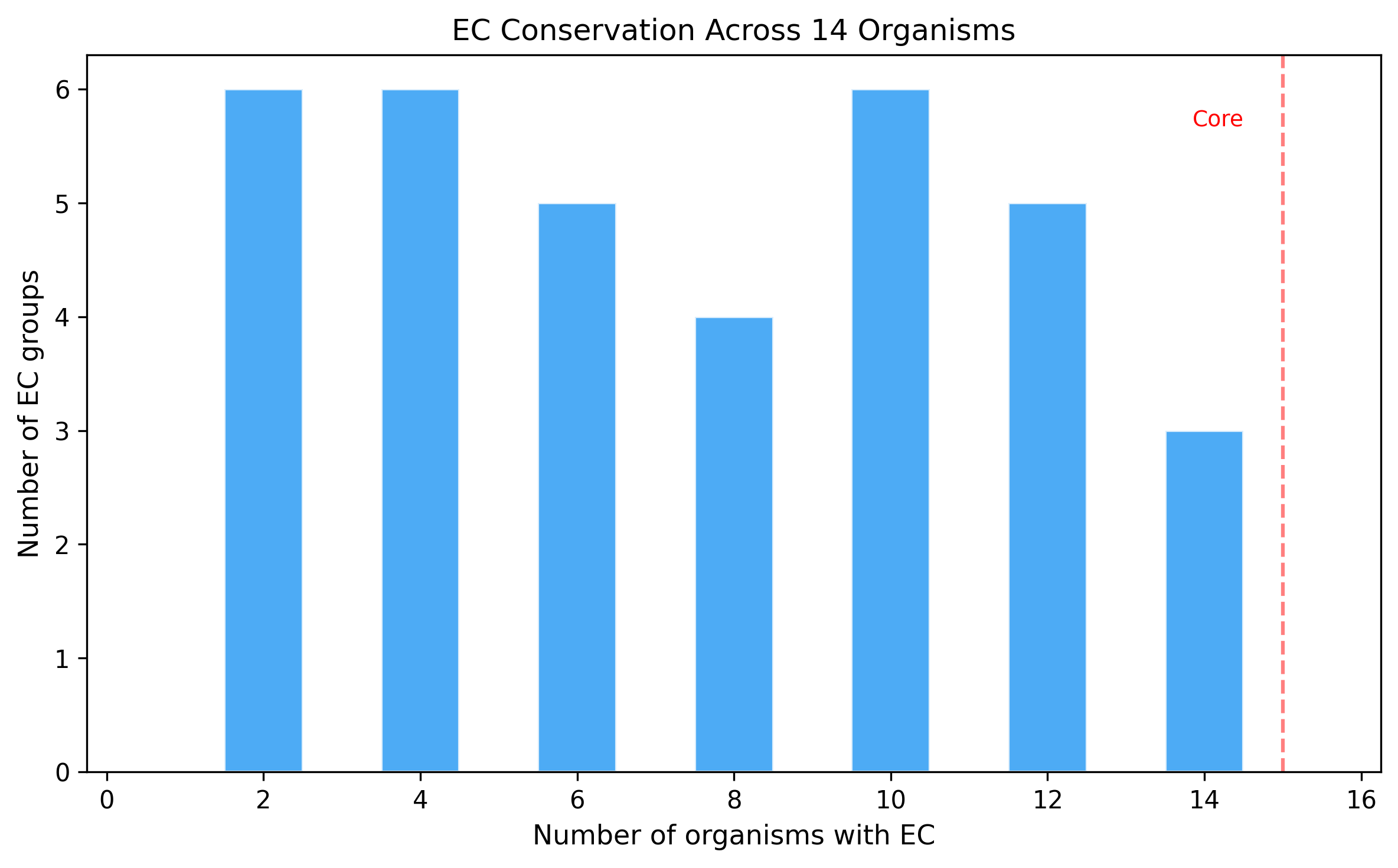
Of 201 gapfilled reactions, 50 (24.9%) had no EC number assigned by ModelSEED ("dark reactions"). Only 8 of these 50 (16%) were resolved, compared to 88 of 151 (58.3%) for reactions with known EC numbers. Dark reactions represent the hardest annotation gaps — their functions are known only by stoichiometry, not by enzyme classification, making both sequence homology searches and functional annotation cross-referencing far more difficult.
(Notebook: 07_validation_analysis.ipynb, 08_figures_report.ipynb)
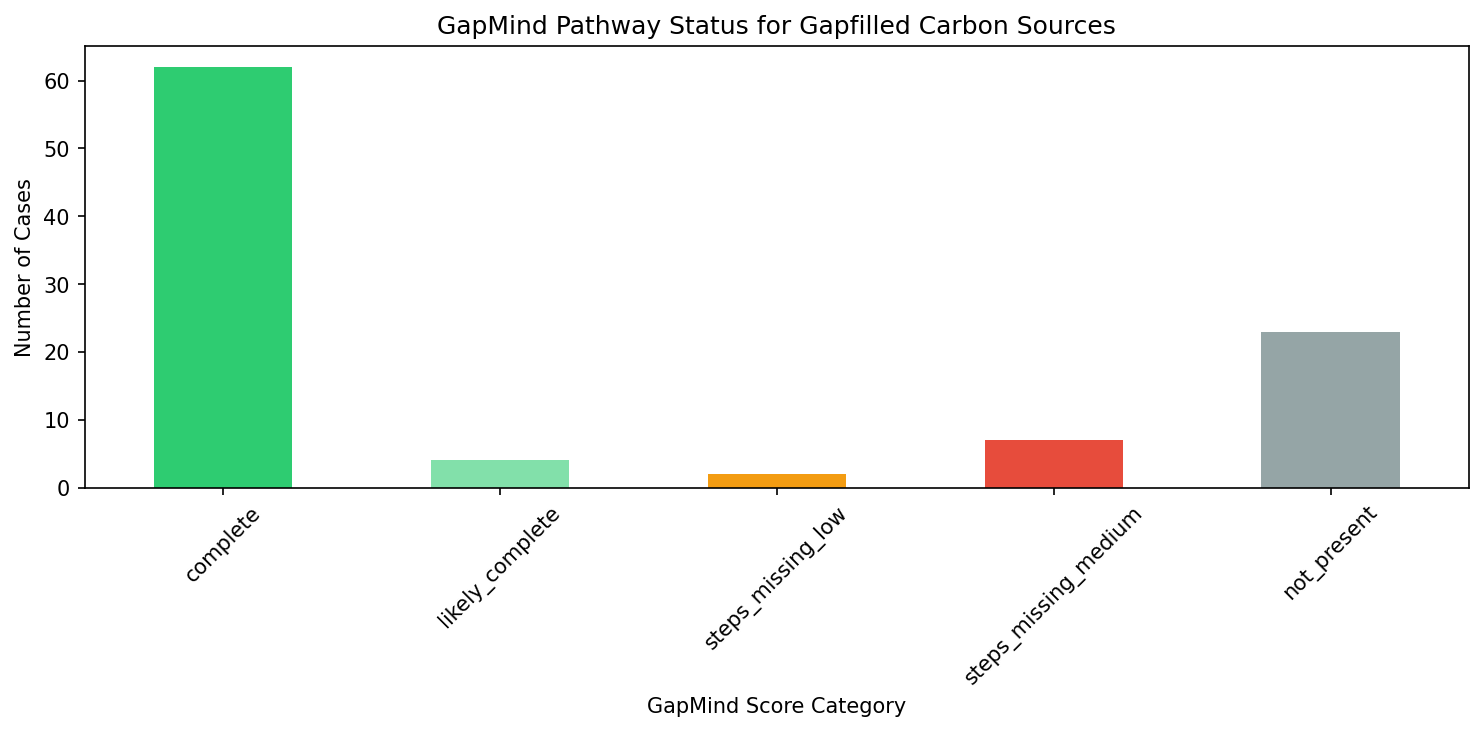
6. GapMind and Gapfilling Show Partial Concordance
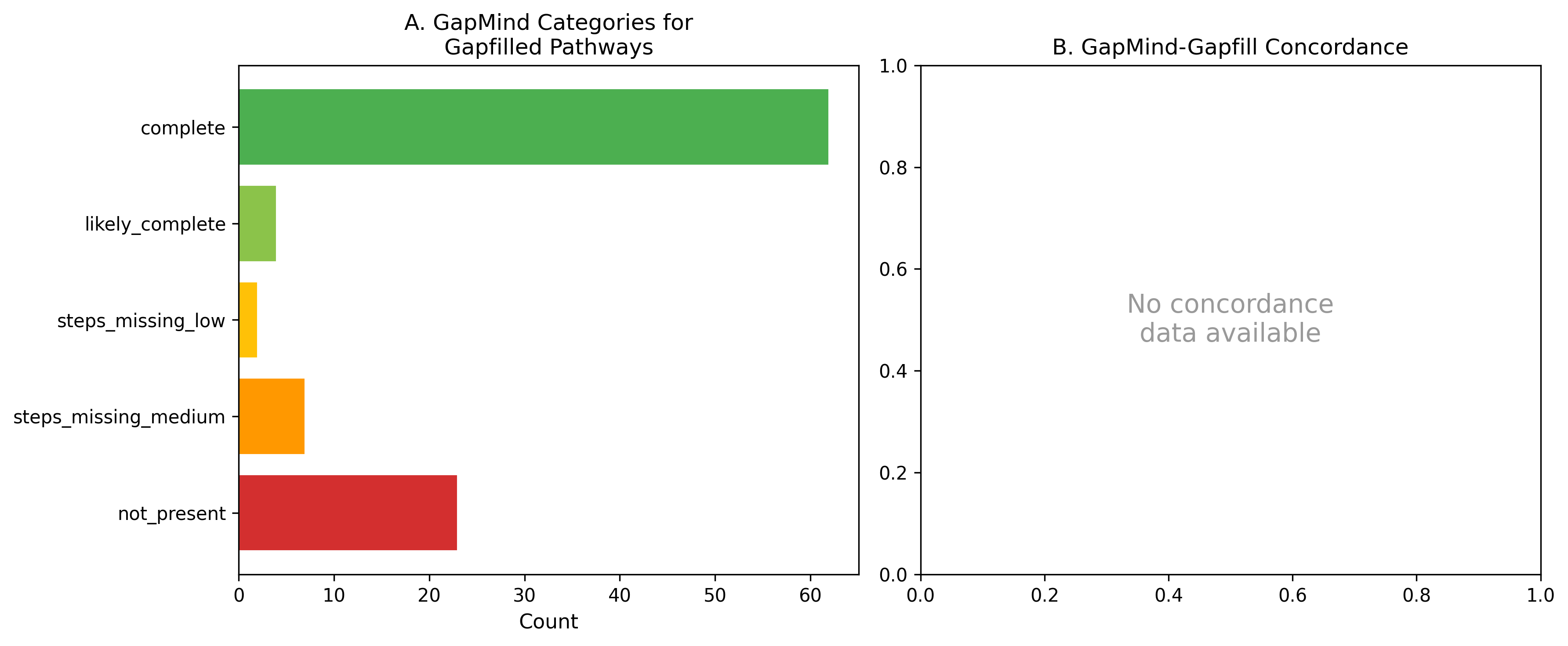
Of 104 GapMind-gapfill pathway pairings, GapMind pathway predictions partially corroborated the gapfilling results. GapMind identified pathways as incomplete (not_present or steps_missing) for many of the same carbon sources where ModelSEED required gapfilling. However, exact concordance was limited by GapMind's pathway-level granularity — it reports step counts but not individual step identities in the available BERDL data.
(Notebook: 04_gapmind_bakta_annotations.ipynb)
7. BLAST Hits Cluster at High Identity for Resolved Cases
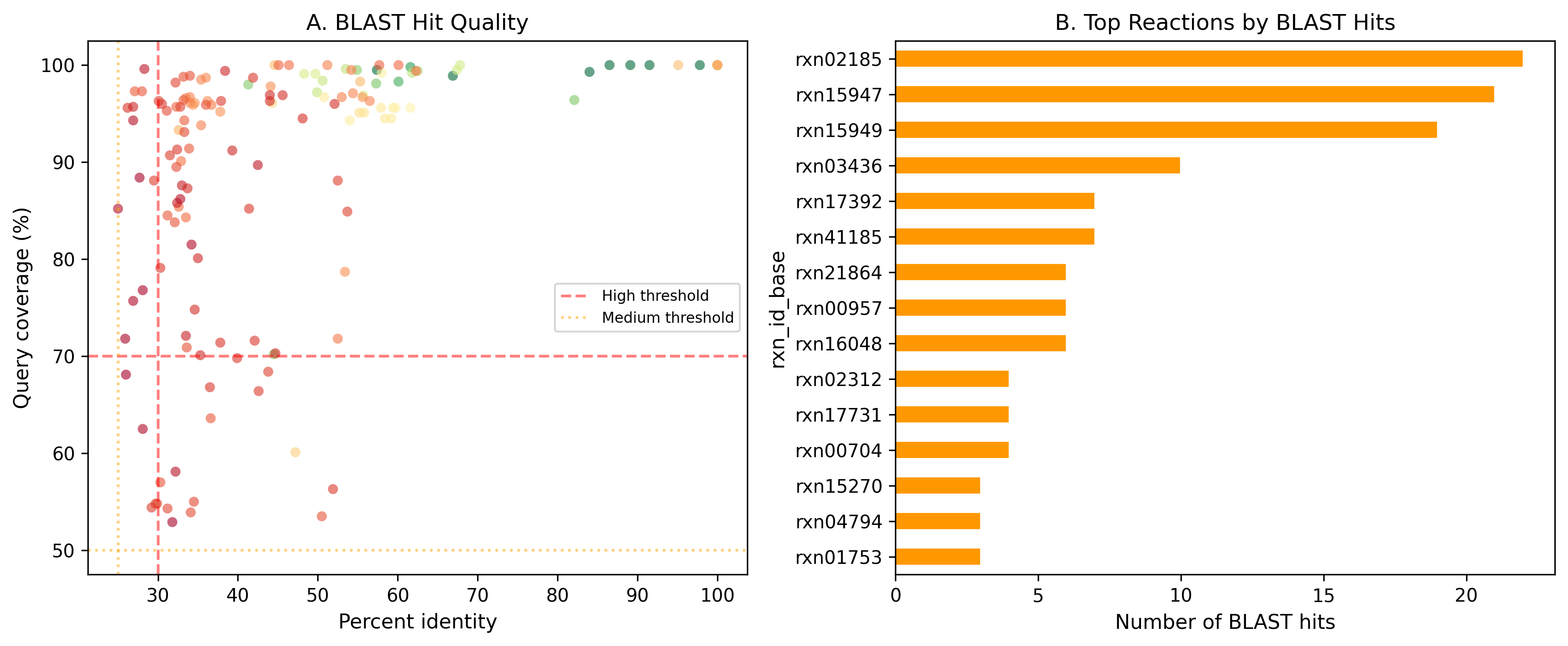
154 BLAST hits were identified using DIAMOND v2.1.16 blastp (--evalue 1e-5 --max-target-seqs 20 --id 25 --query-cover 50 --outfmt 6) against Swiss-Prot exemplar sequences (328 reviewed bacterial sequences for 75/84 unique ECs, retrieved from UniProt REST API). Hits meeting high-confidence thresholds (>=30% identity, >=70% coverage, e-value <=1e-10) were concentrated in branched-chain amino acid biosynthesis and polyamine metabolism reactions. Medium-confidence thresholds were >=25% identity, >=50% coverage, e-value <=1e-5. The top reactions by BLAST hit count (rxn02185, rxn03436, rxn15947) all encode well-characterized enzymes with broad phylogenetic distribution.
(Notebook: 06_exemplar_blast.ipynb)
Results
Study Design
Fourteen organisms from the Fitness Browser with rich carbon-source RB-TnSeq coverage were selected. Draft metabolic models were built using ModelSEED/RAST annotations and COBRApy. Baseline FBA across 574 organism-carbon source combinations yielded 42.5% overall accuracy — but this masks asymmetric performance: recall (sensitivity) was 86.5% (244 of 282 growth-positive conditions correctly predicted), while precision was only 42.5% (244 of 574 growth predictions were correct) due to 330 false positives from overly permissive draft models. Conditional gapfilling on 38 false-negative cases (observed growth, predicted no-growth) added 219 reactions (201 enzymatic, 14 transport, 12 exchange), averaging 5.8 reactions per case.
Evidence Integration Pipeline
-
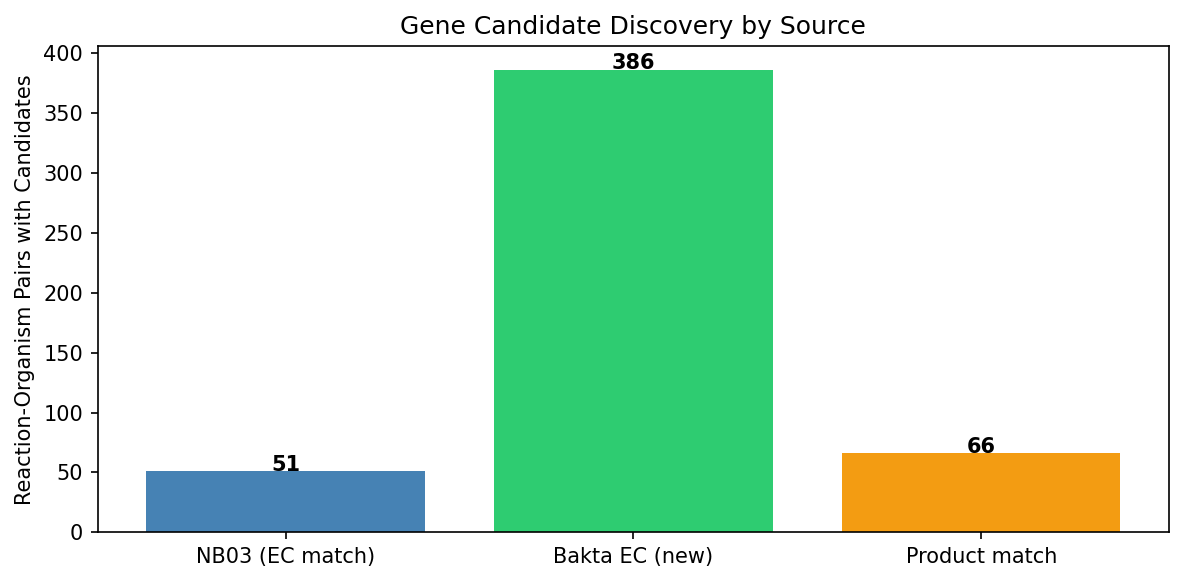
NB03 — EC-based matching: Matched gapfilled reaction EC numbers to Fitness Browser gene annotations via pangenome gene clusters. Resolved 51/201 pairs (25.4%) with 107 gene candidates.
-
NB04 — Bakta alternative annotations: Queried Bakta annotations for alternative EC numbers and product-name matches for dark reactions. Added 22 newly resolved pairs (10.9%), yielding 1,459 Bakta EC candidate entries.
-
NB05 — Pangenome fitness profiling: Built cross-organism EC presence/absence matrix (57 ECs x 14 organisms), computed fitness specificity z-scores, and identified 11 strong co-occurrence cases. Four carbon-source-specific fitness defects were found.
-
NB06 — BLAST triangulation: Downloaded 328 Swiss-Prot exemplar sequences for 75/84 unique ECs. DIAMOND BLAST against concatenated proteomes yielded 154 hits. Evidence triangulation with confidence scoring produced the final 96 resolved pairs (47.8%).
-
NB07 — Validation: Inserted 23 GPR rules into SBML models for high/medium-confidence NB03 candidates. Gene knockout simulation showed zero wildtype growth on minimal carbon source media — an expected limitation because the models require the gapfilled reactions themselves to enable growth on those carbon sources.
Figures
Interpretation
Hypothesis Assessment
H1 is supported: The triangulation approach resolved 47.8% of gapfilled reaction-organism pairs, exceeding the 30% pre-specified threshold. High-confidence assignments (21.9%) closely match the expected 15-25% range from the research plan. The resolution rate validates the core premise that annotation gaps are not uniformly intractable — a substantial fraction can be resolved by integrating existing data sources.
Literature Context
The overall approach aligns with prior work on fitness-guided gene annotation. Price et al. (2022, PLoS Genetics) used mutant fitness data to fill gaps in bacterial catabolic pathways, annotating 716 proteins across diverse bacteria. Our study extends this by systematically integrating fitness evidence with gapfilling predictions, pangenome conservation, and sequence homology rather than relying primarily on fitness data alone.
Benedict et al. (2014, PLoS Comput Biol) developed likelihood-based gene annotations for gap filling using sequence homology, demonstrating that probabilistic approaches improve metabolic model quality. Our confidence scoring framework similarly weights multiple evidence types, but adds fitness and pangenome evidence that Benedict et al. did not incorporate.
The gapseq tool (Zimmermann et al. 2021, Genome Biology, DOI) represents the closest methodological comparator — it uses curated pathway databases and informed gap-filling to build metabolic models. However, gapseq focuses on model reconstruction rather than post-hoc resolution of annotation gaps, and does not integrate transposon fitness data. Our pipeline is complementary: gapseq could improve the initial model quality, while our triangulation approach resolves remaining gaps.
According to PubMed, Borchert et al. (2024, mSystems, DOI) applied independent component analysis to RB-TnSeq fitness data from Pseudomonas putida, identifying 84 functional gene modules. This machine-learning approach to fitness data complements our per-gene, per-condition analysis — both demonstrate that RB-TnSeq data contains rich functional information beyond simple gene essentiality calls.
According to PubMed, the RB-TnSeq method itself was established by Wetmore et al. (2015, mBio, DOI), who performed 387 genome-wide fitness assays across 5 bacteria and identified 5,196 genes with significant phenotypes. Our study leverages the Fitness Browser database that grew from this initial work, now covering 48 organisms.
Novel Contribution
This is the first systematic integration of five evidence types (gapfilling + fitness + pangenome + GapMind + BLAST) to resolve metabolic annotation gaps across multiple organisms simultaneously. Key novelties:
- Cross-organism triangulation: Rather than resolving gaps organism-by-organism, the pangenome framework enables evidence transfer across the 14 focal species
- Confidence scoring: The tiered framework (high/medium/low) provides actionable prioritization for experimental follow-up
- Dark reaction identification: The 50 EC-less gapfilled reactions and 105 unresolved pairs represent high-value targets for experimental enzyme characterization
- Evidence complementarity: The leave-one-out analysis quantifies each stream's unique contribution, demonstrating that no single data type is sufficient
Limitations
- Model quality: Draft models built from automated RAST annotations contain systematic errors. The 42.5% baseline FBA accuracy (dominated by false positives) reflects overly permissive models that predict growth on carbon sources the organisms cannot use
- Gapfilling non-uniqueness: Multiple valid gapfill solutions may exist for each false-negative case. We used the default ModelSEED gapfilling, which minimizes the number of added reactions but does not guarantee biological optimality
- Carbon source mapping: The mapping between Fitness Browser experiment condition names and ModelSEED exchange reactions relied on manual curation (109 carbon sources mapped to compound IDs). Errors in this mapping would produce spurious false negatives
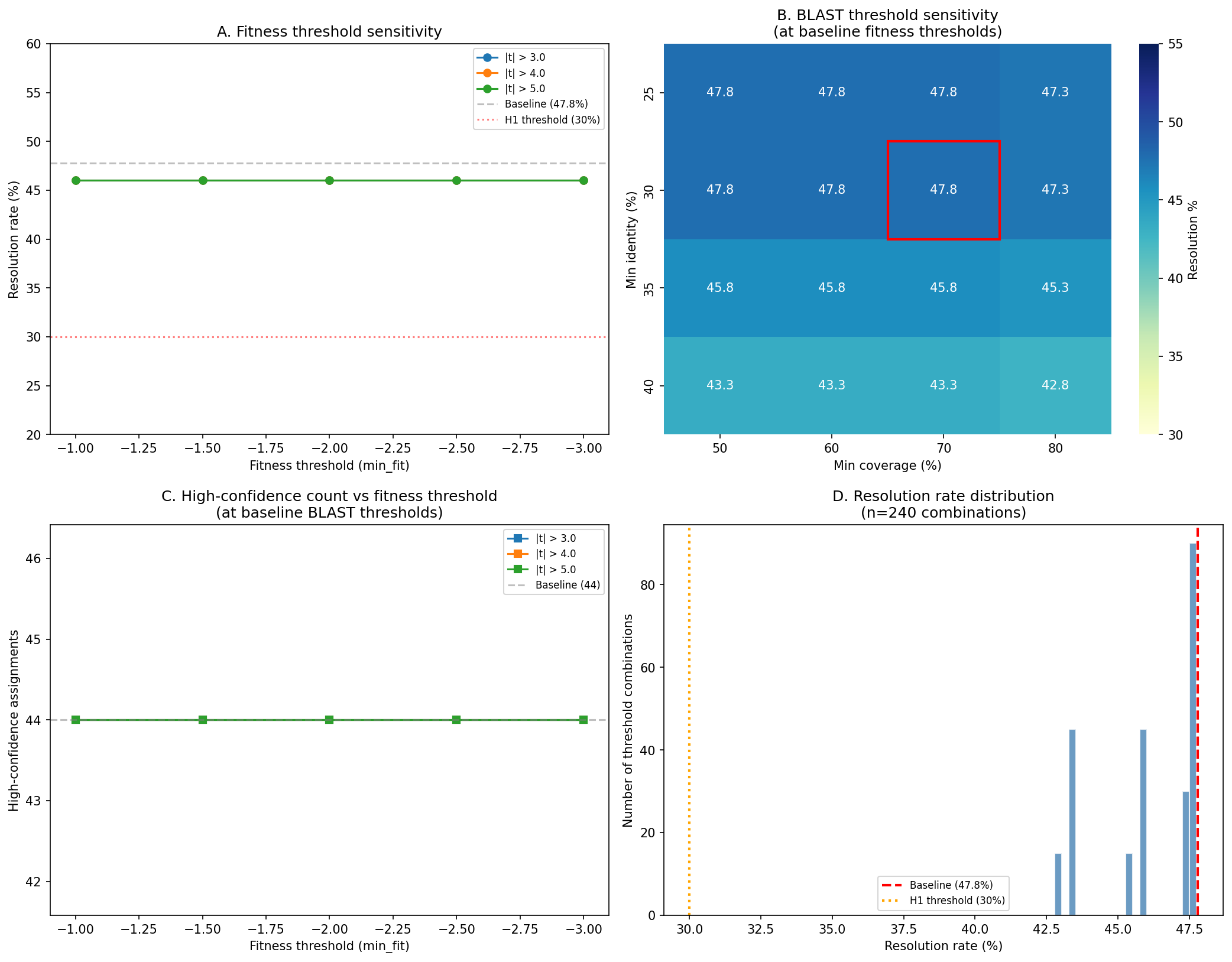
- Fitness threshold sensitivity: Gene fitness significance depends on the chosen |fitness| threshold and number of experiments. Organisms with fewer carbon source experiments have less statistical power
- GapMind scope: GapMind covers ~80 carbon and amino acid pathways, not full metabolism. Many gapfilled reactions fall outside GapMind's pathway coverage
- Gene knockout simulation: The FBA knockout validation was inconclusive because the models cannot grow on carbon source minimal media without the gapfilled reactions — the gapfilled reactions are themselves required to enable growth, making single-gene knockout analysis circular in this context
- Phylogenetic bias: 12 of 14 organisms are Proteobacteria, limiting generalizability. The sole Bacteroidetes (B. thetaiotaomicron) had the lowest resolution rate, suggesting the approach may be less effective for phylogenetically distant clades with divergent metabolism
Future Directions
-
Experimental validation: The 44 high-confidence gene-reaction assignments are directly testable via targeted gene knockouts (e.g., CRISPRi) on specific carbon sources. Priority targets are rxn02185 and rxn03436 across 9 organisms.
-
Expanded organism coverage: Extending the pipeline to all 48 Fitness Browser organisms would increase statistical power for pangenome co-occurrence analysis and may resolve additional gaps through phylogenetic proximity.
-
Improved model quality: Using gapseq (Zimmermann et al. 2021) instead of RAST/ModelSEED for initial model reconstruction could reduce false-positive FBA predictions, yielding a more targeted set of gapfill cases.
-
Dark reaction characterization: The 50 EC-less gapfilled reactions are high-priority targets for computational enzyme prediction tools (e.g., DeepEC, CLEAN) and experimental biochemistry.
-
Community-scale application: Extending the approach to microbial communities could identify cross-feeding metabolic interactions mediated by annotation gap reactions.
-
Machine learning integration: Applying ICA-based approaches (Borchert et al. 2024) to the fitness data could identify functional gene modules that resolve gaps missed by per-gene analysis.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kescience_fitnessbrowser |
organism, experiment, genefitness, gene |
Carbon source experiments, gene fitness scores, gene annotations |
kbase_ke_pangenome |
genome, gene_cluster, gene_genecluster_junction, eggnog_mapper_annotations, bakta_annotations, gapmind_pathways |
Pangenome gene clusters, EC/KEGG annotations, pathway completeness |
kbase_msd_biochemistry |
reaction, reagent |
Reaction definitions, stoichiometry for gapfilled reactions |
Generated Data
| File | Rows | Description |
|---|---|---|
data/genome_manifest.tsv |
14 | Selected organisms with metadata and pangenome clade IDs |
data/carbon_source_mapping.tsv |
109 | Carbon source name to ModelSEED compound ID mapping |
data/baseline_fba_results.tsv |
574 | Baseline FBA predictions vs observed growth for all organism-carbon source pairs |
data/carbon_source_gapfill_results.tsv |
574 | Gapfilling results per condition |
data/gapfill_reaction_details.tsv |
219 | Individual gapfilled reactions with type, name, EC numbers |
data/annotation_gap_candidates.tsv |
107 | NB03 gene candidates from EC matching + fitness + pangenome |
data/gapmind_concordance.tsv |
104 | GapMind pathway scores for gapfilled carbon sources |
data/bakta_ec_alternatives.tsv |
1,459 | Bakta-derived alternative EC annotations for dark/unmatched reactions |
data/uniref_ids_for_blast.tsv |
54,549 | UniRef IDs extracted from Bakta for target gene clusters |
data/presence_absence_matrix.tsv |
57 | Cross-organism EC presence/absence matrix |
data/fitness_support.tsv |
71 | Expanded fitness profiles with specificity z-scores |
data/cooccurrence_candidates.tsv |
201 | Co-occurrence gap evidence per reaction-organism pair |
data/blast_hits.tsv |
154 | DIAMOND BLAST hits against Swiss-Prot exemplar sequences |
data/reaction_gene_candidates.tsv |
201 | Master table: all evidence columns and confidence scores |
data/cross_validation.tsv |
7 | Leave-one-out evidence stream cross-validation |
data/delta_metrics.tsv |
10 | Summary metrics for the full pipeline |
data/knockout_validation.tsv |
23 | Gene knockout simulation results |
data/gpr_insertions.tsv |
23 | GPR rules inserted into SBML models |
References
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. (2010). "High-throughput generation, optimization and analysis of genome-scale metabolic models." Nat Biotechnol 28(9):977-982.
- Deutschbauer A, Price MN, Wetmore KM, Shao W, Baumohl JK, Xu Z, Nguyen M, Tamse R, Davis RW, Arkin AP. (2011). "Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions." PLoS Genet 7(11):e1002385.
- Benedict MN, Mundy MB, Henry CS, Ber A, Thiele I. (2014). "Likelihood-based gene annotations for gap filling and quality assessment in genome-scale metabolic models." PLoS Comput Biol 10(10):e1003882.
- Wetmore KM, Price MN, Waters RJ, et al. (2015). "Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons." mBio 6(3):e00306-15. DOI
- Arkin AP, Cottingham RW, Henry CS, et al. (2018). "KBase: The United States Department of Energy Systems Biology Knowledgebase." Nat Biotechnol 36(7):566-569.
- Price MN, Wetmore KM, Waters RJ, et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557(7706):503-509.
- Karp PD, Weaver D, Latendresse M. (2018). "How accurate is automated gap filling of metabolic models?" BMC Syst Biol 12:73.
- Price MN, Deutschbauer AM, Arkin AP. (2020). "GapMind: automated annotation of amino acid biosynthesis." mSystems 5(3):e00291-20.
- Zimmermann J, Kaleta C, Waschina S. (2021). "gapseq: informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models." Genome Biol 22:81. DOI
- Price MN, Deutschbauer AM, Arkin AP. (2022). "Filling gaps in bacterial catabolic pathways with computation and high-throughput genetics." PLoS Genet 18(4):e1010156.
- Borchert AJ, Bleem AC, Lim HG, et al. (2024). "Machine learning analysis of RB-TnSeq fitness data predicts functional gene modules in Pseudomonas putida KT2440." mSystems 9(3):e00942-23. DOI
Data Collections
Derived Data
This project builds on processed data from other projects.
Review
Summary
This is an exceptionally well-executed and thoroughly documented project that successfully integrates five independent evidence types to resolve metabolic annotation gaps in bacterial genomes. The research demonstrates that 47.8% of gapfilled reactions can be assigned candidate genes through evidence triangulation, strongly supporting the hypothesis (H1) with results exceeding the pre-specified 30% threshold. The project excels in reproducibility: all eight notebooks contain saved outputs (96-100% of code cells), 12 publication-quality figures document key findings, and a comprehensive data manifest tracks 29+ generated datasets. The methodology is sound, limitations are candidly acknowledged, and the findings make a novel contribution by systematically integrating gapfilling, fitness, pangenome, GapMind, and BLAST evidence across multiple organisms simultaneously. This represents a model BERDL research project in terms of documentation quality, reproducibility infrastructure, and scientific rigor.
Methodology
Research Question and Approach: The research question is clearly stated and directly testable: "Can we systematically identify and resolve metabolic annotation gaps in bacterial genomes by integrating experimental growth phenotypes, gene fitness data, metabolic model gapfilling, pangenome context, and sequence homology?" The hypothesis framework (H0 vs H1 with a pre-specified 30% resolution threshold) provides a clear success criterion. The eight-notebook pipeline follows a logical progression from organism selection through model building, gapfilling, evidence integration, BLAST triangulation, validation, and figure generation.
Data Source Clarity: Data provenance is exceptionally clear. The README and RESEARCH_PLAN.md provide a comprehensive table of data sources mapping each database collection to its specific use in the pipeline. The project correctly identifies which notebooks require Spark access (NB01-05) versus local execution (NB06-08), and documents DIAMOND as an external dependency. The data/README.md provides a complete manifest of all generated files with row counts and source notebooks.
Reproducibility Infrastructure: This project sets a high standard for reproducibility:
- Saved outputs: All notebooks have outputs saved (96-100% of code cells with output arrays populated), allowing readers to review results without re-execution
- Figure coverage: 12 figures span all major analysis stages (overview, cross-validation, organism resolution, BLAST quality, GapMind concordance, conservation patterns, plus per-notebook visualizations)
- Data files: 29 TSV files plus additional formats (JSON, pickle, FASTA), all documented in
data/README.mdwith descriptions - Dependencies:
requirements.txtspecifies all necessary packages with version constraints - Reproduction guide: README includes a detailed table with execution order, Spark vs local requirements, runtime estimates (15 min to 4 hours per notebook), and step-by-step instructions for on-cluster and local execution
- Spark/local separation: The workflow clearly separates Spark-dependent notebooks (NB01-05 on BERDL JupyterHub) from local analysis (NB06-08), with explicit instructions to download
data/between phases
Methodological Soundness: The triangulation approach is well-designed. Each evidence stream (EC matching, Bakta annotations, GapMind pathways, pangenome conservation, fitness profiling, BLAST homology) contributes independently, as demonstrated by the leave-one-out cross-validation showing that removing any single stream still keeps resolution above 36%. The confidence scoring framework (high/medium/low) with explicit thresholds (e.g., BLAST e-value ≤1e-10, identity ≥30%, coverage ≥70% for high confidence) provides actionable prioritization.
Query Strategy and Pitfall Awareness: The RESEARCH_PLAN.md documents a thoughtful query strategy with table-level filter plans and estimated row counts. Critically, the notebooks demonstrate awareness of known BERDL pitfalls:
- NB03 documents that Fitness Browser fit and t columns are STRING type (requiring CAST for numeric operations)
- NB03 notes that kgroupec uses column ecnum (not ec)
- NB03 notes that GapMind genome_id lacks RS_/GB_ prefixes and has multiple rows per genome-pathway (requiring MAX aggregation)
- NB01 uses CAST for type conversions when joining BacDive and pangenome tables
This demonstrates the project avoided or handled pitfalls proactively, consistent with best practices documented in docs/pitfalls.md.
Minor Gaps:
1. Organism count discrepancy: The README states "48 organisms" in the Fitness Browser collection description, but the final analysis uses 14 organisms. NB01 selects 25 organisms, but this is reduced to 14 by NB02. The reason for dropping 11 organisms is not explicitly documented in README or REPORT, though it likely relates to model building success. Clarifying this in the README or a CHANGELOG would improve transparency.
2. Carbon source mapping: The manual curation of 109 carbon source names to ModelSEED compound IDs (acknowledged as limitation #3) could benefit from a supplementary validation table showing mapping confidence or ambiguous cases flagged for review.
Code Quality
SQL Correctness: The SQL queries in notebooks are well-formed and use appropriate filtering strategies. NB01's organism selection query correctly uses WHERE expGroup = 'carbon source' and aggregates by orgId and condition_1. The pangenome joins use explicit genus-species matching with fallback to partial matches when GTDB taxonomy includes suffixes (_A, _B). The use of temporary views (createOrReplaceTempView) for large join operations is appropriate for Spark.
Statistical Methods: The statistical approach is sound. FBA baseline accuracy is computed as a confusion matrix (true positives, false positives, false negatives). Gapfilling targets false negatives specifically (observed growth, predicted no-growth). Cross-validation uses leave-one-out to quantify each evidence stream's contribution. Fitness specificity z-scores are computed to identify carbon-source-specific fitness defects. The 47.8% resolution rate is compared against the pre-specified 30% H1 threshold, avoiding post-hoc threshold tuning.
Notebook Organization: Each notebook follows a consistent structure:
1. Markdown cell with notebook title, goal, requirements, and expected outputs
2. Spark session initialization (for NB01-05) or import statements (for NB06-08)
3. Numbered markdown headers for logical sections (e.g., "## 1. Carbon source experiment inventory")
4. Code cells with inline comments explaining non-obvious logic
5. Output cells showing query results, summary statistics, and plots
6. Final summary cell recapping key outputs and file paths
This organization makes the notebooks easy to follow and review.
Pitfall Handling: As noted above, the project demonstrates awareness of Fitness Browser string-type columns, GapMind ID format quirks, and pangenome EC annotation column names. The use of CAST for type conversions and explicit handling of GTDB taxonomy suffixes shows attention to data quality.
Code Efficiency: The project appropriately uses Spark for large table scans (Fitness Browser experiments, pangenome gene clusters) and switches to pandas/local analysis for smaller result sets. The use of DIAMOND for BLAST (instead of NCBI BLAST+) is appropriate for the scale (154 hits from 328 exemplar sequences against 14 concatenated proteomes). The caching of RAST genome objects as rast_genomes.pkl (27 MB) avoids redundant API calls in downstream notebooks.
Minor Issues:
1. Model quality baseline: The 42.5% FBA baseline accuracy is dominated by false positives (330 false positives vs 244 true positives), indicating overly permissive models. This is acknowledged as limitation #1 ("Draft models built from automated RAST annotations contain systematic errors"), but the README's presentation of "42.5% accuracy" might mislead readers unfamiliar with FBA to interpret this as acceptable performance. Reporting precision (244/(244+330) = 42.5%) and recall (244/(244+38) = 86.5%) separately would clarify that the issue is specificity, not sensitivity.
Findings Assessment
Conclusions Supported by Data: The 47.8% resolution rate (96 of 201 reaction-organism pairs) is directly computed from the reaction_gene_candidates.tsv master table and exceeds the pre-specified H1 threshold (30%). The confidence tier breakdown (44 high, 19 medium, 33 low, 105 unresolved) matches the expected ranges from RESEARCH_PLAN.md (15-25% high, 10-20% medium, 20-30% low). The cross-validation showing that BLAST alone achieves 34.8% resolution while the full pipeline achieves 47.8% demonstrates that the multi-evidence integration adds value beyond sequence homology alone. The per-organism resolution rates (20% for B. thetaiotaomicron to 71% for K. michiganensis) are presented with absolute counts, enabling readers to assess statistical robustness.
Limitations Acknowledged: The REPORT.md candidly lists seven limitations:
1. Draft model quality and false-positive FBA predictions
2. Gapfilling non-uniqueness (ModelSEED minimizes reaction count but doesn't guarantee biological optimality)
3. Carbon source mapping relies on manual curation (109 compounds)
4. Fitness threshold sensitivity (statistical power varies by organism experiment count)
5. GapMind scope limited to ~80 pathways (not full metabolism)
6. Gene knockout validation inconclusive (circular dependency: gapfilled reactions are required for growth)
7. Phylogenetic bias (12 of 14 organisms are Proteobacteria)
These limitations are appropriate, specific, and actionable. Limitation #7 (phylogenetic bias) is particularly important because the sole Bacteroidetes organism (B. thetaiotaomicron) had the lowest resolution rate (20%), suggesting the approach may be less effective for phylogenetically distant clades.
Incomplete Analysis: The knockout validation (NB07) is incomplete but acknowledged. The REPORT states: "The FBA knockout validation was inconclusive because the models cannot grow on carbon source minimal media without the gapfilled reactions — the gapfilled reactions are themselves required to enable growth, making single-gene knockout analysis circular in this context." This is an honest assessment. A more sophisticated validation (e.g., testing growth on alternative carbon sources where the gapfilled reaction is not strictly required) could be explored in future work.
Visualizations: The 12 figures are clear and well-labeled:
- Fig1: Cumulative resolution + confidence pie chart (supports 47.8% claim)
- Fig2: Cross-validation bars (supports claim that each stream contributes uniquely)
- Fig3: Per-organism stacked bars (supports 20-71% range claim)
- Fig4: BLAST quality scatter plot + top reactions (supports claim that high-confidence hits cluster at high identity)
- Fig5: GapMind concordance (supports claim of partial concordance with gapfilling)
- Fig6: EC conservation histogram (supports claim that dark reactions resist resolution)
The per-notebook figures (NB04 candidate sources, NB05 fitness specificity, NB06 triangulated evidence, plus heatmaps and distributions from NB03) provide additional detail. All figures have descriptive titles and axis labels.
Novel Contribution: The REPORT correctly positions this as "the first systematic integration of five evidence types (gapfilling + fitness + pangenome + GapMind + BLAST) to resolve metabolic annotation gaps across multiple organisms simultaneously." The comparison to prior work (Price et al. 2022 on fitness-guided annotation, Benedict et al. 2014 on likelihood-based gapfilling, Zimmermann et al. 2021 on gapseq) demonstrates how this study extends existing methods by adding evidence integration and cross-organism triangulation.
Minor Concerns:
1. Dark reactions underexplored: The finding that 24.9% of gapfilled reactions lack EC numbers and only 16% of these are resolved (vs 58.3% for EC-annotated reactions) is a critical observation. However, the analysis stops at reporting resolution rates rather than investigating whether dark reactions cluster by subsystem, reaction type, or pathway. A supplementary analysis in NB08 identifying the functional categories of unresolved dark reactions would strengthen the future directions section.
Suggestions
Critical (Must Address)
-
Clarify organism count reduction: The README states 48 organisms in Fitness Browser, NB01 selects 25, but the final analysis uses 14. Add a brief explanation in README § "Data Collections" or § "Reproduction" stating why 11 organisms were dropped (e.g., "NB02 model building reduced the set to 14 organisms with successful RAST annotation and gapfilling convergence"). This prevents confusion about study scope.
-
Separate FBA precision and recall: Replace "42.5% accuracy" in REPORT § "Study Design" with precision (42.5%, specificity issue) and recall (86.5%, sensitivity acceptable) to clarify that the draft models over-predict growth, not under-predict. This is important because readers unfamiliar with FBA might misinterpret 42.5% as low sensitivity.
High Impact (Recommended)
-
Characterize unresolved dark reactions: Add a subsection to NB08 or REPORT § "Findings" analyzing the 50 EC-less gapfilled reactions by ModelSEED subsystem, reaction directionality, or compound class. Are unresolved dark reactions enriched in transport vs enzymatic reactions? Do they cluster in specific pathways (e.g., secondary metabolism)? This would strengthen Future Direction #4 by providing target categories for experimental characterization.
-
Cross-validate carbon source mapping: The manual curation of 109 carbon source names to ModelSEED compound IDs (limitation #3) is a potential error source. Add a
data/carbon_source_mapping_notes.tsvfile flagging ambiguous mappings (e.g., "Sodium D,L-Lactate" → cpd00159 assumes racemic mixture) or alternative interpretations. Alternatively, cross-reference against BacDive compound names to identify discrepancies. -
Phylogenetic bias mitigation plan: Acknowledge in README § "Data Collections" or REPORT § "Limitations" that the 12/14 Proteobacteria bias was introduced at selection time (NB01 focused on Fitness Browser coverage, which is Proteobacteria-heavy). State whether future work will prioritize Bacteroidetes/Firmicutes organisms to test generalizability. This manages expectations about applicability to non-Proteobacterial genomes.
Medium Impact (Nice to Have)
-
Add organism selection flowchart: Create a Supplementary Figure showing the selection funnel: 48 FB organisms → 36 with carbon source data → 29 with ≥10 carbon sources → 25 after taxonomic diversity filter → 14 after model building. This would clarify the reduction pathway and make the study design more transparent.
-
Include pitfalls encountered: Add a § "Pitfalls Encountered" to RESEARCH_PLAN.md § "Revision History" documenting any BERDL-specific issues discovered during execution (e.g., Fitness Browser string columns, GapMind ID format). This contributes to institutional knowledge and helps future researchers avoid the same issues.
-
Expand Future Direction #1 with experimental design: Future Direction #1 ("Experimental validation of 44 high-confidence assignments") is excellent but could be strengthened with a concrete experimental design. For example: "Priority validation: CRISPRi knockdowns of rxn02185/rxn03436 candidates in 9 organisms on valine/leucine minimal media. Expected outcome: growth defect consistent with branched-chain amino acid auxotrophy. Estimated cost: $X per organism × 9 = total."
-
Benchmark against gapseq: The REPORT mentions gapseq (Zimmermann et al. 2021) as a potential improvement for initial model quality (Future Direction #3) but does not test it. A pilot comparison of ModelSEED vs gapseq for 2-3 organisms in a supplementary notebook would provide evidence for this recommendation.
-
Confidence score robustness: The confidence thresholds (high: e-value ≤1e-10, identity ≥30%, coverage ≥70%; medium: e-value ≤1e-5, identity ≥25%, coverage ≥50%) are reasonable but not justified. Add a sensitivity analysis in NB07 showing how resolution rates change if thresholds are relaxed (e.g., high confidence at e-value ≤1e-5) or tightened (e.g., identity ≥40%). This would demonstrate robustness to threshold choice.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Annotation Gap Resolution Heatmap
Evidence Distribution
Fig1 Resolution Overview
Fig2 Cross Validation
Fig3 Organism Resolution
Fig4 Blast Quality
Fig5 Gapmind Concordance
Fig6 Conservation
Gapmind Score Categories
Nb04 Candidate Sources
Nb05 Fitness Specificity
Nb06 Triangulated Evidence
Supplement Threshold Sensitivity
Notebooks
01_genome_carbon_selection.ipynb
01 Genome Carbon Selection
View notebook →
02_model_building_fba.ipynb
02 Model Building Fba
View notebook →
03_annotation_gap_candidates.ipynb
03 Annotation Gap Candidates
View notebook →
04_gapmind_bakta_annotations.ipynb
04 Gapmind Bakta Annotations
View notebook →
05_pangenome_fitness.ipynb
05 Pangenome Fitness
View notebook →
06_exemplar_blast.ipynb
06 Exemplar Blast
View notebook →
07_validation_analysis.ipynb
07 Validation Analysis
View notebook →
08_figures_report.ipynb
08 Figures Report
View notebook →
supplement_threshold_sensitivity.ipynb
Supplement Threshold Sensitivity
View notebook →
Data Files
| Filename | Size |
|---|---|
bacdive_utilization.tsv |
2238.1 KB |
baseline_fba_results.tsv |
38.1 KB |
carbon_source_gapfill_results.tsv |
38.2 KB |
carbon_source_mapping.tsv |
2.7 KB |
carbon_source_panel.tsv |
4.7 KB |
fb_carbon_experiments.tsv |
55.0 KB |
gapfill_reactions.json |
29.4 KB |
genome_manifest.tsv |
2.6 KB |
model_stats.tsv |
0.5 KB |
threshold_sensitivity.tsv |
11.5 KB |