Prophage-AMR Co-mobilization Atlas
CompletedResearch Question
At pangenome scale (293K genomes, 27K species), are antibiotic resistance genes preferentially located within or adjacent to prophage regions, and does this co-localization predict AMR gene mobility and accessory-genome status?
Overview
Five completed AMR projects characterized resistance gene distribution, conservation, fitness costs, and environmental patterns. Separately, the prophage_ecology project mapped prophage distribution across phylogeny. This project connects the two: testing whether AMR genes near prophage markers are more likely to be accessory (mobile) and whether prophage-rich species have broader AMR repertoires. Uses bakta_amr for curated AMR hits, bakta_annotations product-field keywords for prophage marker detection, and gene coordinate proximity for co-localization scoring.
Key Findings
Finding 1: AMR genes frequently share contigs with prophage markers
Over half (55.7%) of AMR gene instances in the top-100 AMR-burdened species reside on contigs that also carry strict prophage markers (terminase, phage structural proteins, holin/lysin). Among these co-localized AMR genes, the median distance to the nearest prophage marker is 34 genes, and 10.4% of all AMR genes are within 10 genes of a prophage marker.
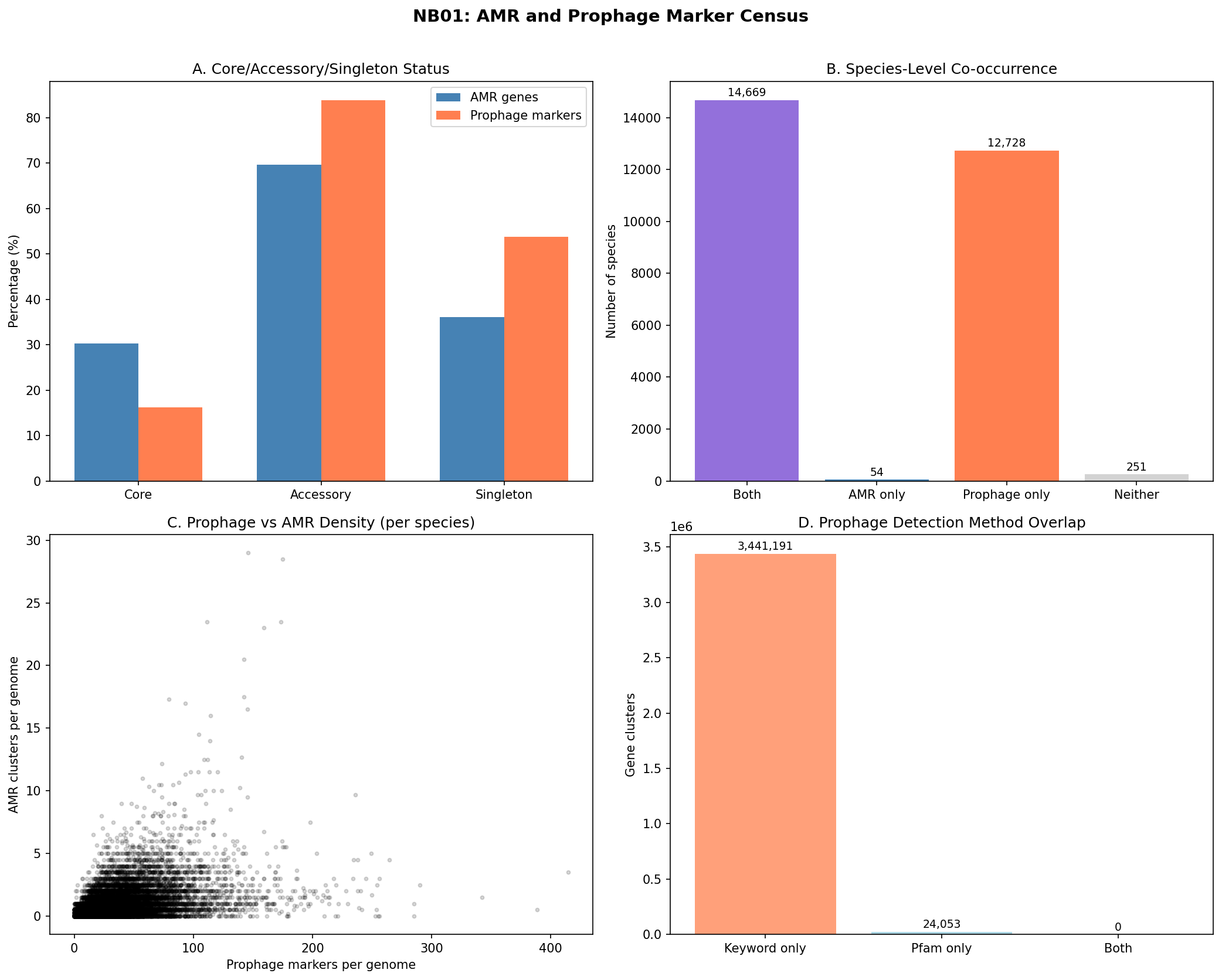
Across the full pangenome inventory, 83,008 AMR gene clusters and 3.47 million prophage marker clusters were identified. Prophage markers are overwhelmingly accessory (83.8%) and frequently singleton (53.8%), while AMR genes are also predominantly accessory (69.7%) but less singleton (36.1%). Of 27,702 species in the pangenome, 14,669 (52.9%) carry both AMR and prophage markers.
(Notebook: 01_amr_prophage_census.py)
Finding 2: Prophage proximity weakly predicts AMR gene mobility (H1)
Prophage-proximal AMR genes (within 10 genes) are slightly more likely to be accessory (67.6%) than distal AMR genes (65.5%), yielding a statistically significant but modest effect (Fisher's exact OR=1.10, p=0.005, bootstrap 95% CI [1.024, 1.185]).
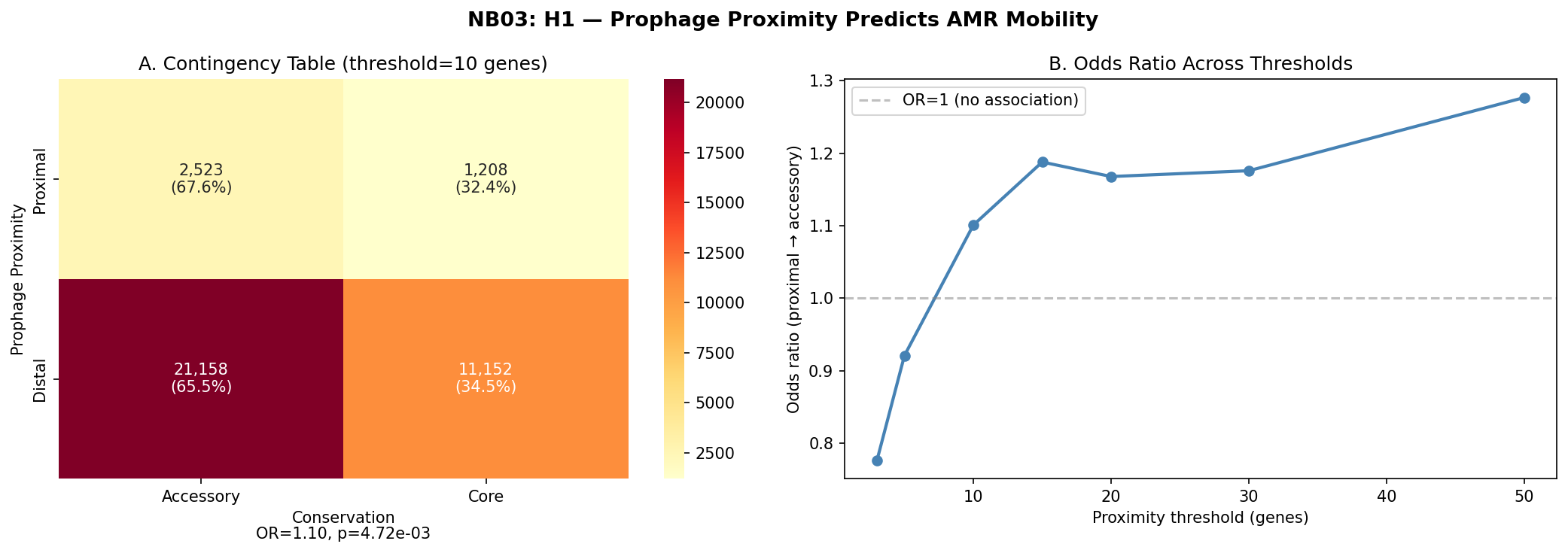
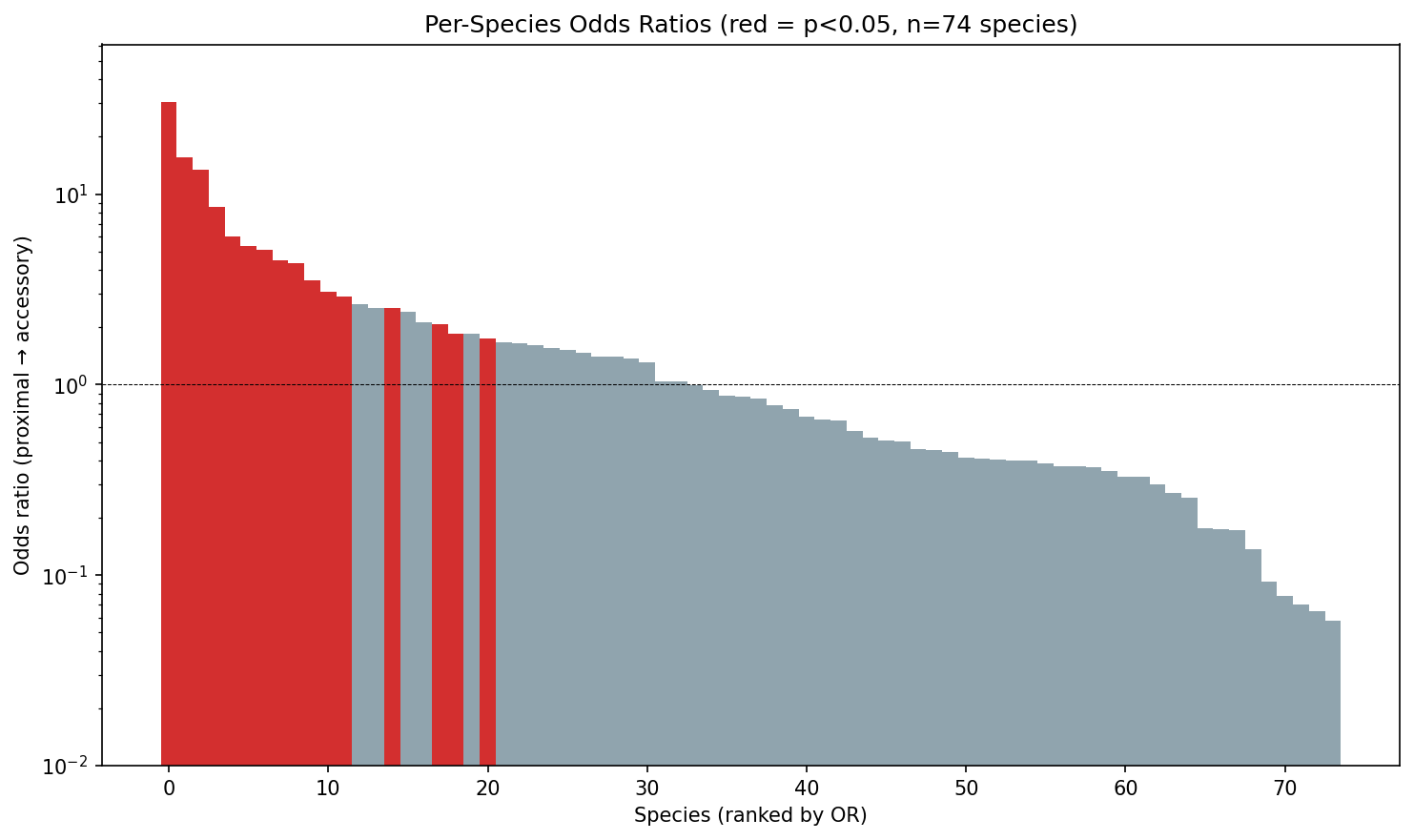
The effect is threshold-dependent: it is absent or reversed at very close range (OR=0.78 at 3 genes, OR=0.92 at 5 genes) and strengthens at broader thresholds (OR=1.19 at 15 genes, OR=1.28 at 50 genes). Per-species analysis reveals heterogeneity: only 33 of 74 testable species show OR>1, with a median species-level OR of 0.85.
(Notebook: 03_conservation_test.py)
Finding 3: Prophage density strongly predicts AMR repertoire breadth (H2)
Species with higher prophage marker density carry significantly broader AMR gene repertoires (Spearman rho=0.572, p<10^-300, n=4,770 species). A log-log regression reveals that a 10-fold increase in prophage density predicts a ~6.6-fold increase in AMR breadth (slope=0.823, R²=0.30). The association is robust after controlling for genome count (partial Spearman rho=0.464, p=1.0×10^-253).
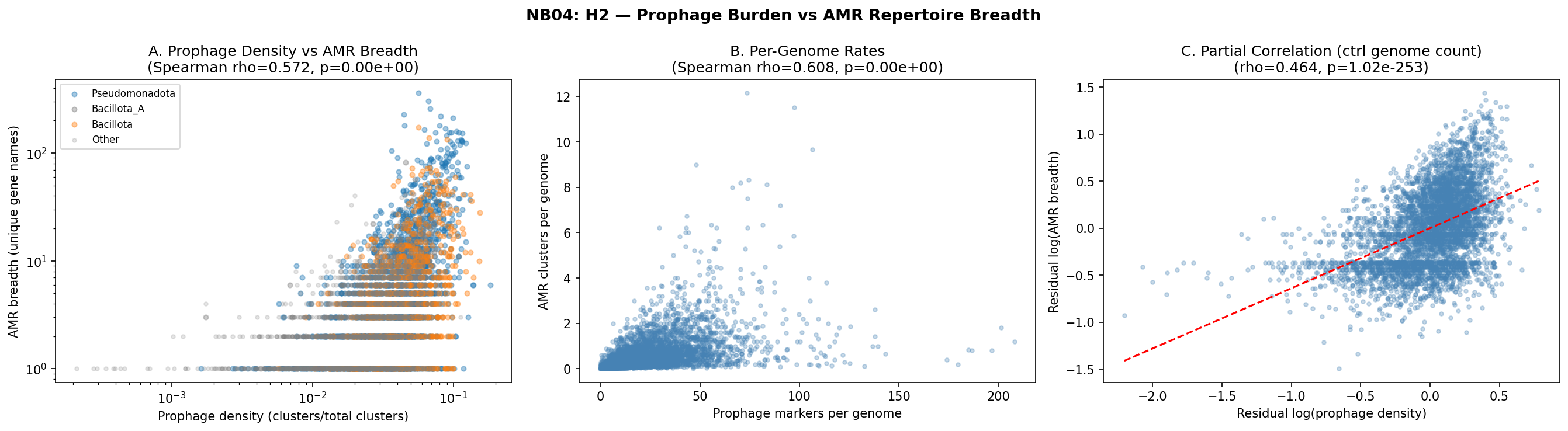
The correlation is significant across all five major phyla: Pseudomonadota (rho=0.54), Bacillota_A (rho=0.55), Bacillota (rho=0.40), Bacteroidota (rho=0.59), and Actinomycetota (rho=0.29). This pan-phylogenetic consistency argues against phylogenetic confounding.
(Notebook: 04_species_breadth_test.py)
Finding 4: Fitness cost comparison not testable (H3)
The BERDL fitness browser covers only 48 model organisms with RB-TnSeq data, which do not overlap well with the GTDB pangenome species analyzed here. H3 could not be tested and remains an open question for future work with expanded fitness datasets.
(Notebook: 05_synthesis.py)
Results
Pangenome-Scale Census
| Metric | Value |
|---|---|
| AMR gene clusters | 83,008 |
| Prophage marker clusters (strict) | 1,261,929 |
| Prophage marker clusters (broad) | 3,465,244 |
| Species with both AMR and prophage | 14,669 |
| AMR clusters that are accessory | 69.7% |
| Prophage clusters that are accessory | 83.8% |
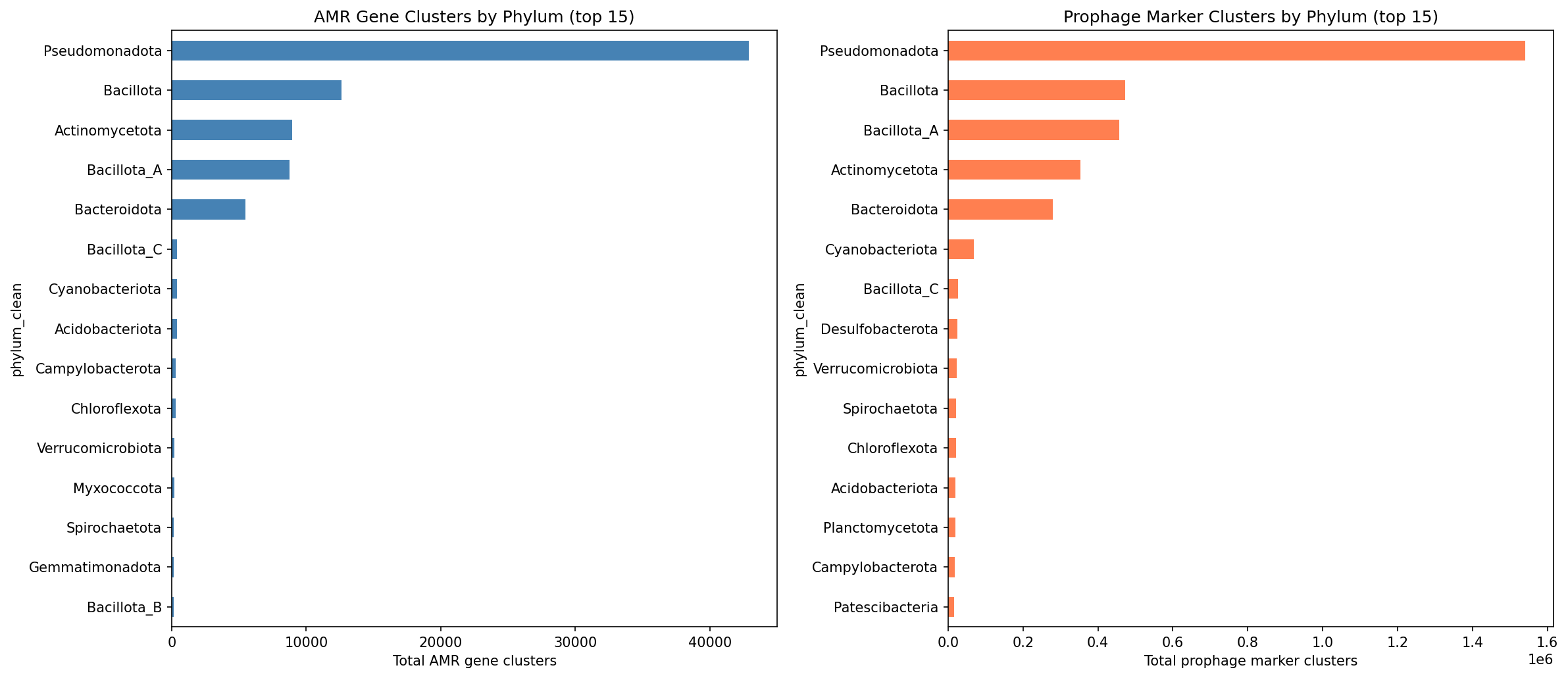
Gene Neighborhood Co-localization
Across 100 species (20 genomes sampled per species, 1,953 genomes total), 36,041 AMR gene instances were analyzed:
| Distance threshold | AMR instances | % of total |
|---|---|---|
| On prophage contig | 20,073 | 55.7% |
| Within 50 genes | 12,026 | 33.4% |
| Within 20 genes | 7,137 | 19.8% |
| Within 10 genes | 3,731 | 10.4% |
| Within 5 genes | 1,991 | 5.5% |
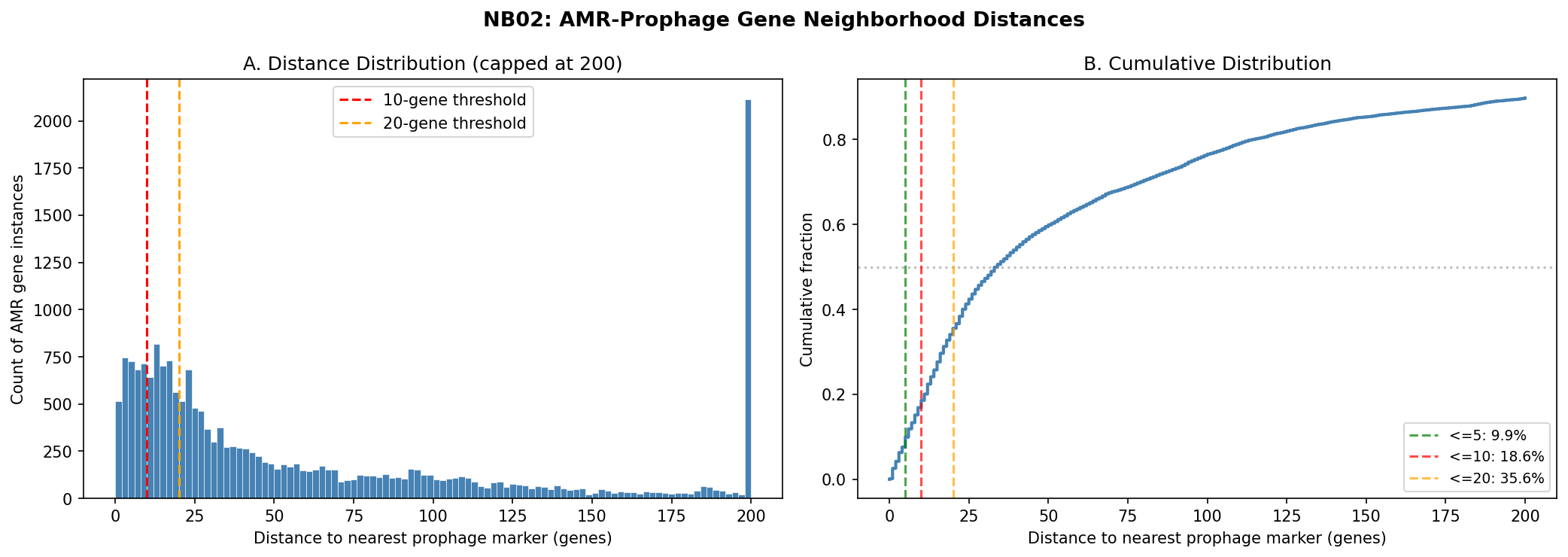
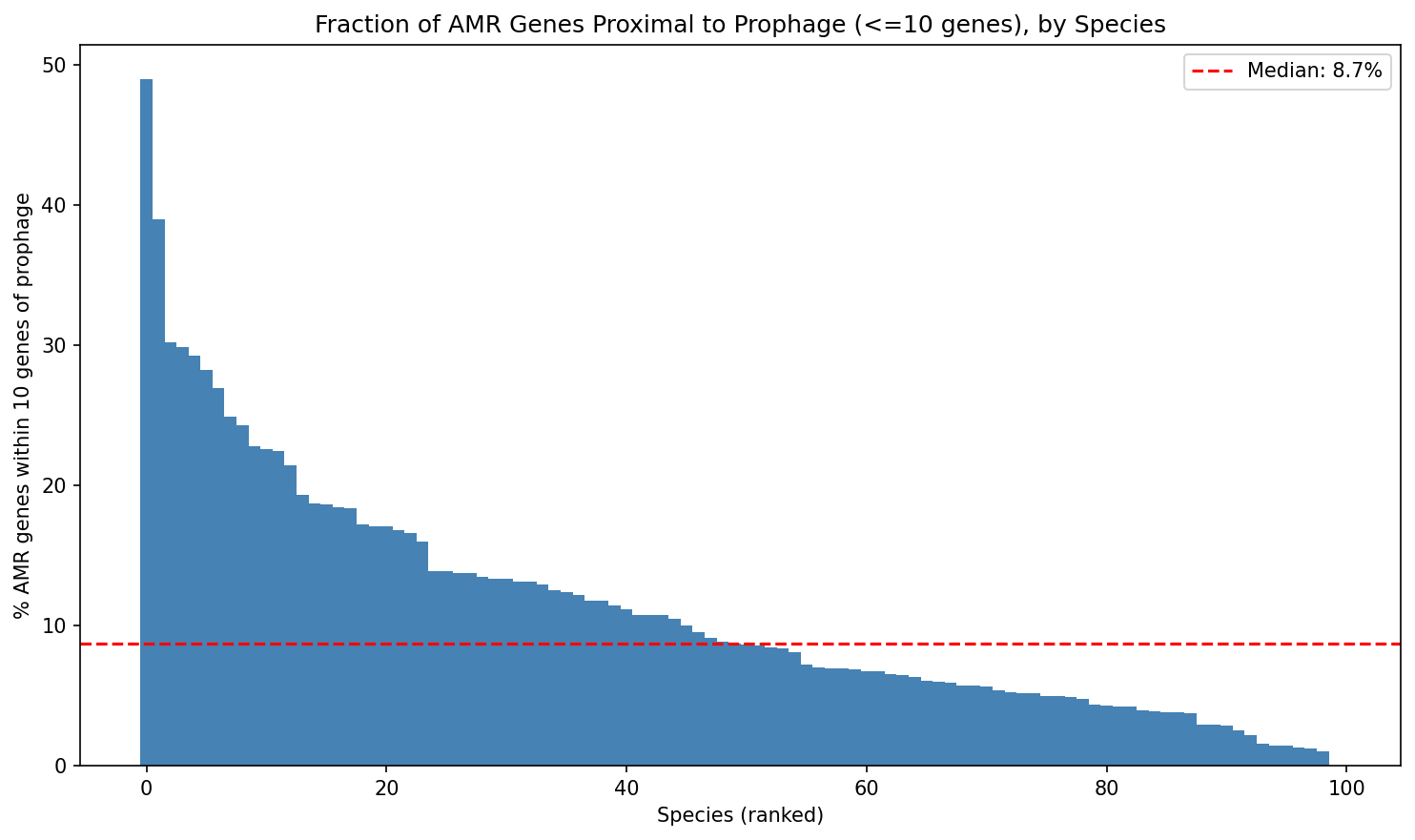
H1: Conservation × Proximity
| Condition | Accessory | Core | % Accessory |
|---|---|---|---|
| Proximal (≤10 genes) | 2,523 | 1,208 | 67.6% |
| Distal (>10 genes) | 21,158 | 11,152 | 65.5% |
Fisher's exact test (one-sided): OR=1.10, p=0.005. Bootstrap 95% CI: [1.024, 1.185].
H2: Species-Level Breadth
| Test | Statistic | p-value |
|---|---|---|
| Spearman: prophage density vs AMR breadth | rho=0.572 | <10^-300 |
| Spearman: prophage/genome vs AMR/genome | rho=0.608 | <10^-300 |
| Log-log regression slope | 0.823 (SE=0.018) | <10^-300 |
| Partial Spearman (ctrl genome count) | rho=0.464 | 1.0×10^-253 |
Interpretation
The strongest finding is that prophage density is a powerful species-level predictor of AMR repertoire breadth (H2). This is consistent with two non-exclusive mechanisms: (1) prophages directly mobilize resistance genes via specialized or generalized transduction, and (2) species with high recombination potential (indicated by many prophages) are also better at acquiring genes from other mobile elements. The effect persists after controlling for genome count and is consistent across all five major phyla, arguing against a purely phylogenetic explanation.
The gene-level co-localization result (H1) is more nuanced. While statistically significant in aggregate, the effect is modest (2.1 percentage point difference in accessory fraction) and heterogeneous across species. The reversal at very close range (3-5 genes) may reflect that genes immediately adjacent to phage structural genes are core phage components rather than recently acquired cargo. The strengthening at broader thresholds (15-50 genes) could capture "genomic islands" that include both phage remnants and laterally transferred genes.
Literature Context
Based on articles retrieved from PubMed:
-
Our H2 finding aligns with Rendueles et al. (2018) (DOI), who showed that bacteria encoding capsules have more prophages AND more antibiotic resistance genes across >100 pangenomes. Our work extends this by demonstrating the prophage-AMR correlation at much larger scale (4,770 species vs ~100) and showing it persists after controlling for genome count rather than capsule presence.
-
Chen et al. (2018) (DOI) found ARG-MGE co-occurrence on assembled contigs from river metagenomes, including associations with prophages. Our pangenome-scale analysis provides a genomic-reference counterpart to their metagenomic observations, confirming that the ARG-prophage association is encoded in reference genomes, not just environmental assemblies.
-
Bearson & Brunelle (2015) (DOI) demonstrated that fluoroquinolone exposure induces prophage in multidrug-resistant Salmonella, enabling phage-mediated transduction of resistance plasmids. This provides a direct mechanistic link supporting our correlative finding.
-
Fisarova et al. (2021) (DOI) showed that S. epidermidis phages transduce antimicrobial resistance plasmids at high frequency, providing additional mechanistic evidence for phage-mediated AMR spread in clinically relevant organisms.
Novel Contribution
- Scale: This is the first pangenome-scale analysis of prophage-AMR gene-neighborhood co-localization across 100 species (1,953 genomes, 36,041 AMR gene instances).
- Species-level predictor: Prophage density explains 30% of variance in AMR breadth across 4,770 species — a stronger predictor than previously documented.
- Threshold sensitivity: The reversal of the proximity-accessory association at very close range (≤5 genes) suggests that immediate phage neighbors are core phage genes, not acquired cargo. This nuance was not captured in prior contig-level studies.
Limitations
- Gene position proxy: Distances are in ordinal gene positions parsed from gene_id format, not base-pair resolution. True genomic distances may differ.
- Prophage identification: Uses keyword/Pfam matching on bakta_annotations rather than dedicated prophage prediction tools (e.g., PHASTER, geNomad). May include false positives (e.g., phage-defense systems) and miss divergent prophages.
- Sampling: 20 genomes sampled per species for co-localization (NB02). While representative, exhaustive analysis of all 293K genomes would strengthen the findings.
- Core/accessory labels: Depend on species-level pangenome calling (motupan). The same gene in different species may have different conservation status.
- Fitness data gap: H3 could not be tested due to limited overlap between fitness browser organisms and GTDB species.
- Correlation vs causation: The H2 association does not prove phage-mediated AMR transfer; species with open pangenomes may independently accumulate both prophages and AMR genes.
Future Directions
- Dedicated prophage prediction: Apply geNomad or PHASTER to BERDL genomes and ingest results as a new table. This would replace keyword-based prophage detection with a validated tool and reduce false positives.
- Base-pair resolution: Use contig sequences (scaffoldseq) to calculate true genomic distances rather than ordinal gene positions.
- Expanded fitness analysis: As fitness browser coverage expands, revisit H3 to test whether prophage-proximal AMR genes have distinct fitness costs.
- Plasmid vs phage partitioning: Distinguish between AMR genes mobilized by prophages vs plasmids vs ICEs using the pangenome-level co-localization data.
- Clinical strain focus: Analyze the WHO priority pathogen subset (K. pneumoniae, A. baumannii, P. aeruginosa, E. coli) in greater depth with all available genomes.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
bakta_amr, bakta_annotations, bakta_pfam_domains |
AMR and prophage marker identification |
kbase_ke_pangenome |
gene, gene_genecluster_junction, gene_cluster |
Gene coordinates and cluster membership |
kbase_ke_pangenome |
genome, pangenome, gtdb_taxonomy_r214v1 |
Species membership, genome counts, taxonomy |
kescience_fitnessbrowser |
genefitness, gene, organism |
Attempted fitness cost comparison (H3) |
Generated Data
| File | Rows | Description |
|---|---|---|
data/amr_clusters.csv |
83,008 | AMR gene clusters with species, core/accessory status, gene names |
data/prophage_marker_clusters.csv |
3,465,244 | Prophage marker clusters from keyword + Pfam detection |
data/amr_prophage_species_summary.csv |
27,702 | Per-species AMR and prophage counts with taxonomy |
data/amr_prophage_distances.csv |
36,041 | Per-AMR-gene-instance distance to nearest prophage marker |
data/census_summary.json |
— | Aggregate census statistics |
data/coloc_summary.json |
— | Co-localization summary statistics |
data/h1_test_results.json |
— | H1 Fisher's exact test results and threshold sensitivity |
data/h2_test_results.json |
— | H2 regression and correlation results |
data/h3_test_results.json |
— | H3 fitness comparison results (not tested) |
data/project_synthesis.json |
— | Integrated synthesis of all hypotheses |
References
- Rendueles O, de Sousa JAM, Bernheim A, Touchon M, Rocha EPC. (2018). "Genetic exchanges are more frequent in bacteria encoding capsules." PLoS Genet. 14(12):e1007862. DOI. PMID: 30576310.
- Chen H, Chen R, Jing L, Bai X, Teng Y. (2018). "A metagenomic analysis framework for characterization of antibiotic resistomes in river environment." Environ Pollut. 245:398-407. DOI. PMID: 30453138.
- Bearson BL, Brunelle BW. (2015). "Fluoroquinolone induction of phage-mediated gene transfer in multidrug-resistant Salmonella." Int J Antimicrob Agents. 46(2):201-4. DOI. PMID: 26078016.
- Fisarova L, et al. (2021). "Staphylococcus epidermidis Phages Transduce Antimicrobial Resistance Plasmids and Mobilize Chromosomal Islands." mSphere. 6(3):e00223-21. DOI. PMID: 33980677.
- Price MN, et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature. 557:503-509. DOI. PMID: 29769716.
- Arkin AP, et al. (2018). "KBase: The United States Department of Energy Systems Biology Knowledgebase." Nat Biotechnol. 36:566-569. DOI. PMID: 29979655.
Data Collections
Atlas Reuse
Derived products, review objects, and tensions connected to this project in the BERIL Atlas.
Review
Summary
This is an exemplary pangenome-scale study investigating the relationship between prophage markers and antibiotic resistance genes across 293K genomes and 27K species. The project successfully demonstrates that prophage density is a strong species-level predictor of AMR repertoire breadth (H2), while gene-level co-localization shows more modest effects (H1). The methodology is sound, the statistical analyses are appropriate, and the findings are well-supported by comprehensive data analysis. The work makes a novel contribution to understanding AMR mobilization mechanisms at unprecedented scale. The main limitation is the use of Python scripts instead of Jupyter notebooks, which reduces immediate reproducibility for readers.
Methodology
The research approach is scientifically rigorous with clearly stated, testable hypotheses. The use of BERDL databases is appropriate, with thoughtful handling of the limitation that genomad_mobile_elements is not available (addressed through keyword + Pfam-domain prophage detection). The statistical methods are well-chosen: Fisher's exact test for H1, Spearman correlation and regression for H2, with proper controls for confounders like genome size and phylogeny.
Data sources are clearly identified and the sampling strategy (top-100 species, 20 genomes per species) balances computational feasibility with representativeness. The threshold sensitivity analysis (3-50 genes) for proximity definition shows methodological sophistication. The authors appropriately acknowledge when H3 could not be tested due to limited fitness data overlap.
Code Quality
The SQL queries are well-structured and appropriate for the BERDL database schema. The Python code demonstrates good practices with clear documentation, proper error handling for different Spark session contexts, and logical organization. Statistical analyses use appropriate libraries (scipy.stats, statsmodels) and methods.
The code appropriately handles known BERDL pitfalls, such as using species-level filtering for large tables and avoiding .toPandas() on large intermediate results. The prophage detection strategy using both bakta_annotations keywords and bakta_pfam_domains is well-implemented with comprehensive pattern matching.
However, there is one significant code organization issue: all notebooks are in Python script format (.py) rather than Jupyter notebook format (.ipynb). While the scripts are well-documented and functional, this means readers cannot see cell-by-cell outputs and must re-run the entire analysis to examine intermediate results.
Findings Assessment
The conclusions are strongly supported by the presented data. The H2 finding (prophage density predicts AMR breadth: Spearman rho=0.572, R²=0.30) is robust and consistent across phylogenetic groups. The H1 result is more nuanced but appropriately interpreted - the modest overall effect (OR=1.10) combined with threshold dependence and species heterogeneity suggests a complex relationship rather than a simple linear association.
The literature context is excellent, positioning findings relative to Rendueles et al. (2018) and Chen et al. (2018), while acknowledging mechanistic support from Bearson & Brunelle (2015) and Fisarova et al. (2021). The authors appropriately distinguish correlation from causation and acknowledge limitations around prophage detection methods and gene position proxies.
Visualizations are comprehensive and well-designed, with 8 figures covering the full analysis pipeline from census through hypothesis testing to synthesis. All key results are supported by both tabular data and graphical presentation.
Suggestions
-
Convert to Jupyter notebook format: The most important improvement would be converting the .py scripts to .ipynb format with saved outputs. This would allow readers to see intermediate results, figures, and data summaries without re-running the computationally intensive Spark queries.
-
Add notebook runtime documentation: Include expected execution times for each notebook in the README reproduction section, noting which require Spark access vs local execution.
-
Expand H3 analysis: Consider testing fitness effects in the subset of organisms where pangenome and fitness data overlap, even if limited. This could provide preliminary insights for the fitness hypothesis.
-
Add prophage detection validation: Compare keyword/Pfam results against a subset analyzed with dedicated prophage prediction tools (PHASTER, geNomad) to estimate false positive/negative rates.
-
Base-pair resolution analysis: For a high-priority subset of species, re-analyze using actual genomic coordinates from scaffold sequences rather than ordinal gene positions.
-
Document sampling representativeness: Add analysis comparing the top-100 species sample to the full species set on key properties (genome size, AMR burden, taxonomy) to demonstrate representativeness.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Nb01 Amr Prophage Phylum Distribution
Nb01 Census Overview
Nb02 Distance Distribution
Nb02 Proximal Fraction
Nb03 H1 Contingency
Nb03 H1 Species Odds
Nb04 H2 Breadth Regression
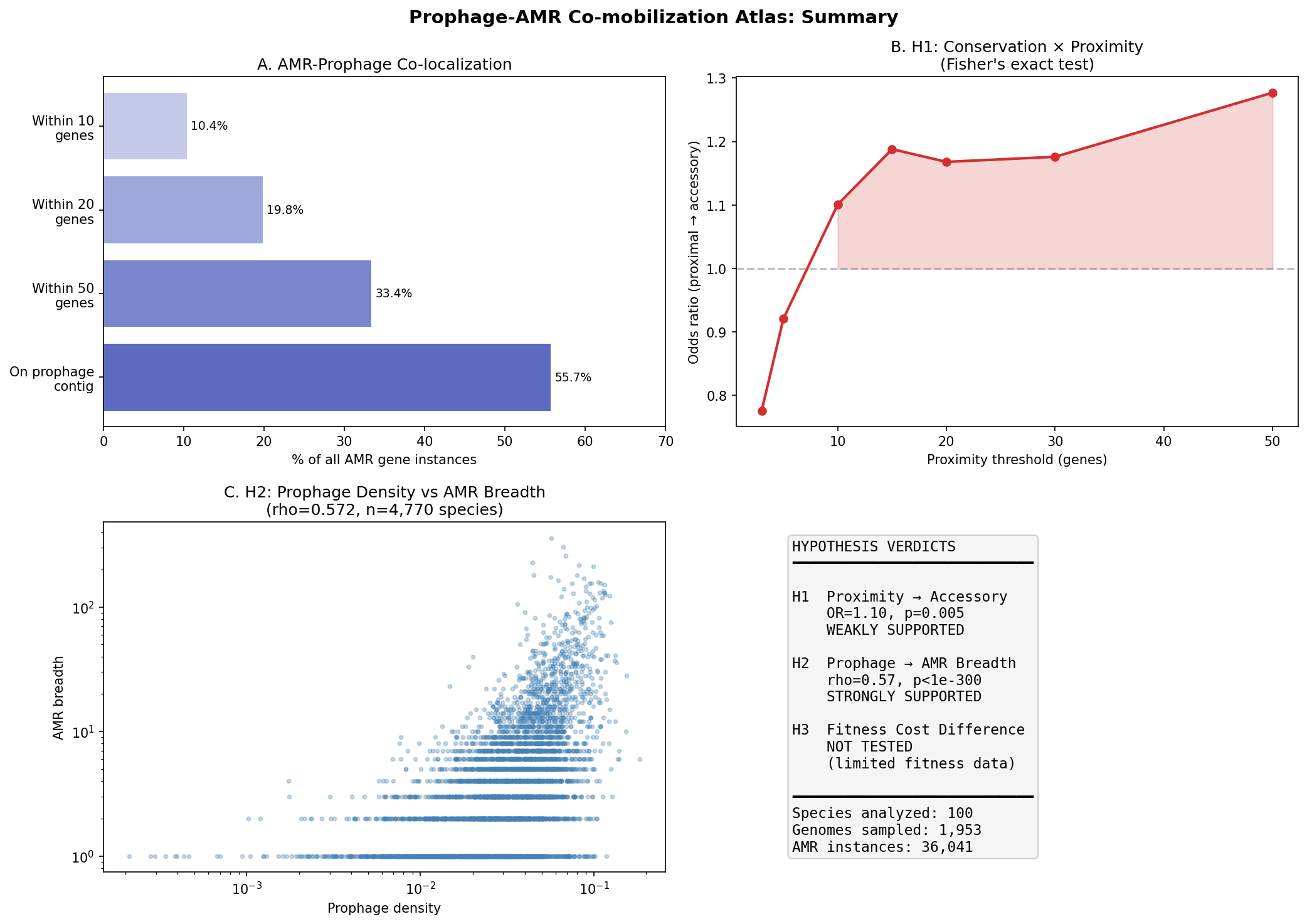
Nb05 Synthesis
Data Files
| Filename | Size |
|---|---|
census_summary.json |
0.7 KB |
coloc_summary.json |
0.4 KB |
h1_test_results.json |
2.1 KB |
h2_test_results.json |
3.5 KB |
h3_test_results.json |
0.2 KB |
project_synthesis.json |
1.8 KB |