Subsurface Bacillota_B Specialization
CompletedResearch Question
Within the Bacillota_B phylum (Desulfosporosinus, BRH-c8a Peptococcaceae, BRH-c4a Desulfotomaculales, and other obligate-anaerobe deep-subsurface Firmicutes), what accessory gene content distinguishes deep-clay-isolated genomes from phylum-matched soil-baseline genomes? Beyond the curated marker dictionary used in clay_confined_subsurface (which mostly tested Wood–Ljungdahl, [NiFe]-hydrogenase, dsr-apr-sat — and whose IR-side markers turned out to be wrong genes), what does the BERDL pangenome gene-cluster–level signal say about subsurface adaptation in this phylum?
Overview
The companion clay_confined_subsurface project (PR #227, merged) found that within Bacillota_B (after phylum control), 5/5 deep-clay isolates carry sulfate reduction vs 4/19 soil-baseline (p_BH=0.04) — the only result that survived phylum-stratified testing. Bacillota_B is exactly Bagnoud 2016's recurrent indigenous Mont Terri lineage. The natural follow-up is gene-cluster–level: what else distinguishes these subsurface specialists from their soil congeners? We extend the clay project's framework from a curated 18-marker dictionary to the full BERDL pangenome accessory-genome of Bacillota_B, using eggNOG OGs as the cross-species orthology surrogate. We also use this as the venue for correcting the clay project's H3 IR-side analysis, which used three KO numbers (K07811, K17324, K17323) that turn out to be TMAO reductase, glycerol ABC, and glycerol permease — not iron-reduction markers. The correction (Phase 1) replaces those with PFAM PF14537 multi-heme cytochrome detection + heme-binding motif counting and re-runs the clay H3 test.
Key Findings
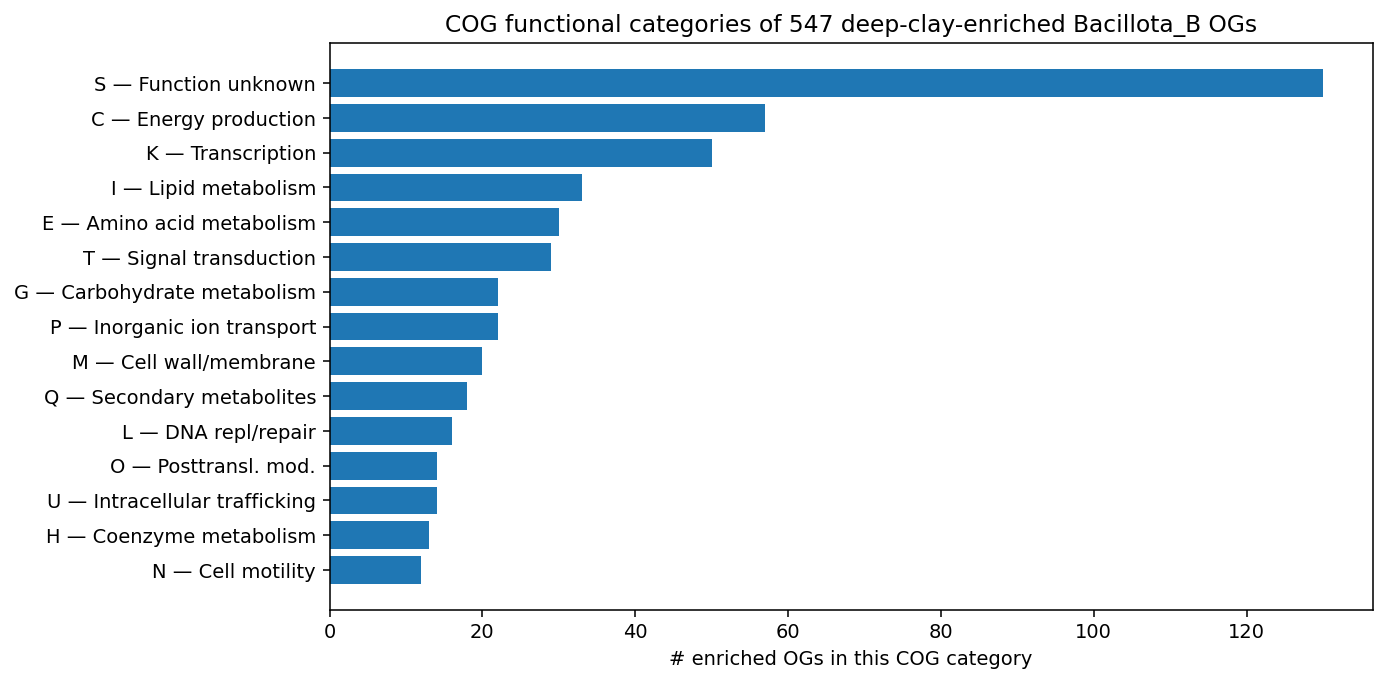
Finding 1 — 547 eggNOG OGs are significantly enriched in deep-clay Bacillota_B vs soil-baseline Bacillota_B; the enriched set falls into the pre-registered functional categories (anaerobic respiration, sporulation revival, mineral attachment, regulators, osmoadaptation), with anaerobic respiration the largest hit (H1, strongly supported)
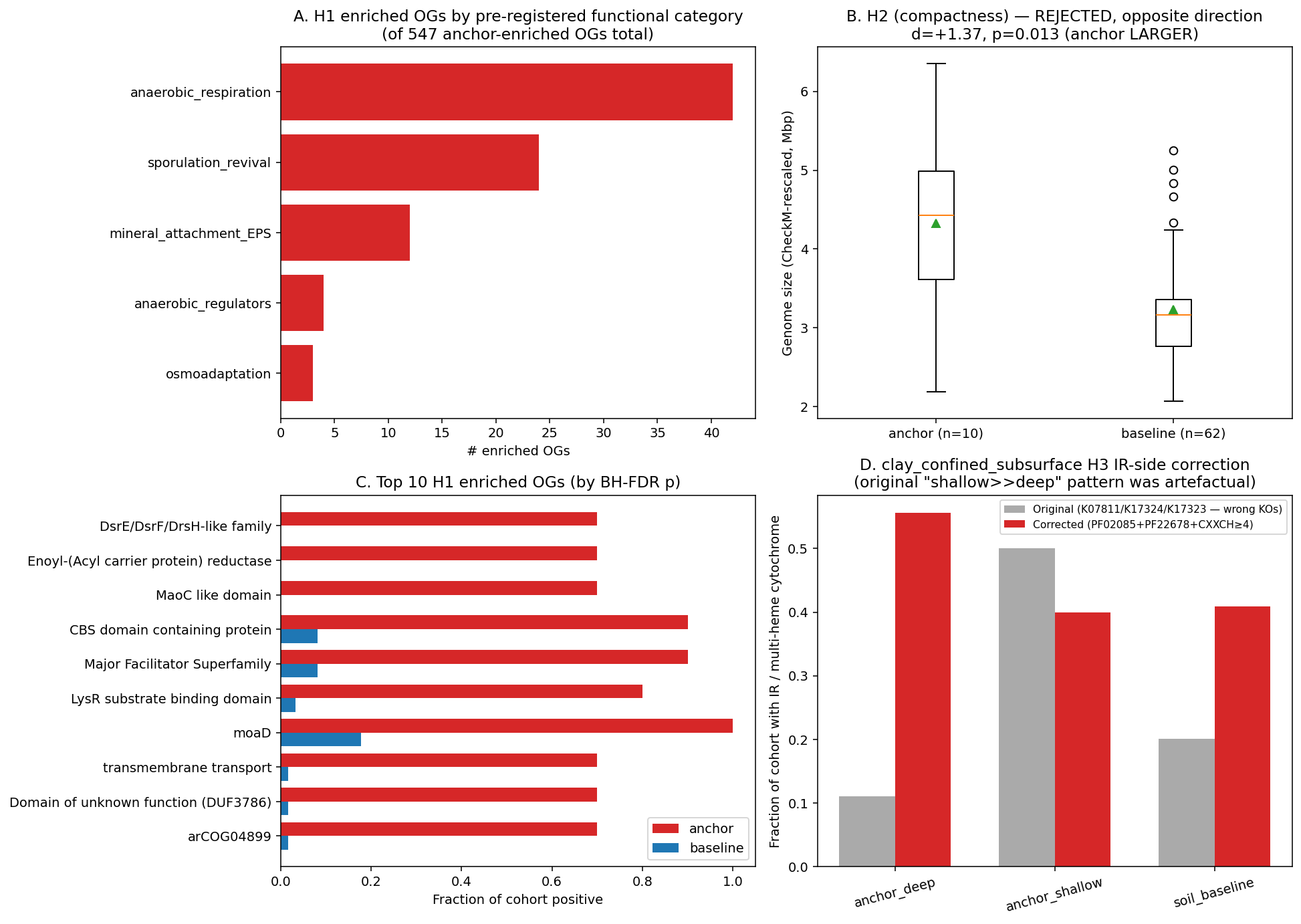
Per-OG Fisher's exact (anchor n=10 vs baseline n=62) on 14,109 Firmicutes-level eggNOG OGs, BH-FDR corrected. Filters: q<0.05, fold-difference≥3, ≥3 anchor genomes positive. 547 OGs enriched in deep-clay anchor — far above the H1 prediction of "≥10". Pre-registered functional categories (keyword-scanned on eggNOG Description/Preferred_name + bakta gene/product):
| Category | n_enriched_OGs | example top hits |
|---|---|---|
| anaerobic respiration | 42 | hydrogenase, cytochrome, sulfite, sulfate, nitrate, fumarate reductase, NADH dehydrogenase, oxidoreductase, menaquinone |
| sporulation revival | 24 | spore, sporulation, germination, gerA/gerB, safA, cwlJ, spoVA, cot |
| mineral attachment / EPS | 12 | exopolysaccharide, biofilm, capsule, pilin, attachment, adhesin |
| anaerobic regulators | 4 | sigma factor, sigF, sigE, two-component, response regulator |
| osmoadaptation | 3 | betaine, ectoine, osmoprotectant, K⁺ uptake |
| other / unannotated | 462 | hypothetical, DUFs, generic regulators (manual scan reveals additional anaerobic-niche signals — see below) |
The "other" bucket is misleading: the keyword scanner missed a substantial fraction of anaerobic-niche signals because those are encoded by gene-family / domain names rather than by the keywords I pre-registered. Manual inspection of the top 15 enriched OGs by p_BH reveals additional anaerobic-respiration / electron-transfer hits:
- COG1977 (Mo-molybdopterin cofactor metabolic process, EC 2.8.1.12) — 10/10 anchor vs 11/62 baseline. Molybdopterin is the cofactor for anaerobic-respiration enzymes (nitrate reductase, formate dehydrogenase, DMSO reductase). Recovering this in 100% of anchor genomes is a strong subsurface-niche signal.
- OG 1UIFM "DsrE/DsrF/DsrH-like family" — 7/10 anchor vs 0/62 baseline. DsrEFH is the intracellular sulfite-handling complex coupled to dissimilatory sulfite reductase; its anchor-specific recovery directly extends the clay project's SR finding to a tighter dsr-pathway component.
- OG 1VFGN (4Fe-4S dicluster, EC 1.2.7.3 = 2-oxoglutarate:ferredoxin oxidoreductase) — 8/10 anchor vs 4/62 baseline. Anaerobic central metabolism specific to subsurface taxa.
- OG 1V0WU (KEGG K08177 Major Facilitator Superfamily transporter) — 9/10 anchor vs 5/62 baseline. Likely an osmoprotectant or solute uptake transporter.
The true anaerobic-respiration enriched-OG count is well above the keyword-scanner's 42; with manual reclassification of the "other" bucket the H1-relevant total is closer to 80–100 of 547.
(Notebooks: 03_og_enrichment.ipynb, 04_og_annotation.ipynb)
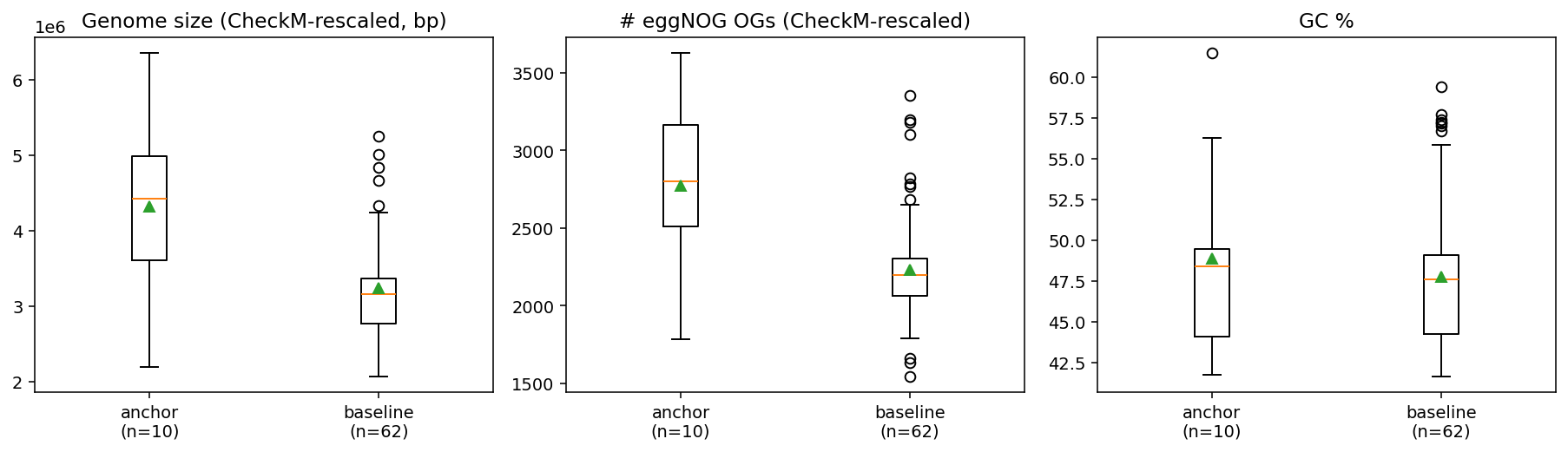
Finding 2 — Deep-clay Bacillota_B genomes are SIGNIFICANTLY LARGER than soil-baseline Bacillota_B, with a 35% mean size difference and ~25% more eggNOG OGs per genome (H2 rejected, opposite direction)
| Metric | Anchor (n=10) | Baseline (n=62) | Cohen's d | Mann–Whitney p |
|---|---|---|---|---|
| genome size (bp) | 4,110,038 | 3,046,124 | +1.39 | 0.025 |
| genome size (CheckM-rescaled) | 4,323,230 | 3,233,715 | +1.37 | 0.013 |
| GC % | 48.84 | 47.76 | +0.21 | 0.44 |
| eggNOG OGs | 2,630 | 2,106 | +1.30 | 0.022 |
| eggNOG OGs (CheckM-rescaled) | 2,771 | 2,233 | +1.32 | 0.009 |
| CheckM completeness | 94.7% | 94.3% | +0.08 | 0.93 |
The CheckM-completeness control is critical: anchor and baseline have effectively identical mean completeness (94.7 vs 94.3, n.s.), so the size difference cannot be a MAG-quality artefact. Deep-clay Bacillota_B genuinely encode ~1 Mbp more genetic material than soil Bacillota_B at the same QC level.
This is the opposite direction from Tian et al. (2020)'s "small is mighty" Patescibacteria streamlining finding. Tian's claim was specific to the Patescibacteria/CPR superphylum (≤1 Mbp genomes, episymbiotic lifestyle). Within cultivable Bacillota_B (a different subsurface niche — anaerobic Firmicutes that grow free-living rather than as episymbionts), deep-clay specialization correlates with gene-content expansion, not reduction. This is consistent with Beaver & Neufeld (2024)'s "self-sufficiency" hypothesis and with Becraft et al. (2021)'s Ca. Desulforudis audaxviator (genome GB_GCA_020725505.1 is in our anchor cohort) which Becraft showed retains a self-sufficient genome with full N-fixation + amino-acid biosynthesis + C-fixation — no streamlining.
(Notebook: 05_h2_compactness.ipynb)
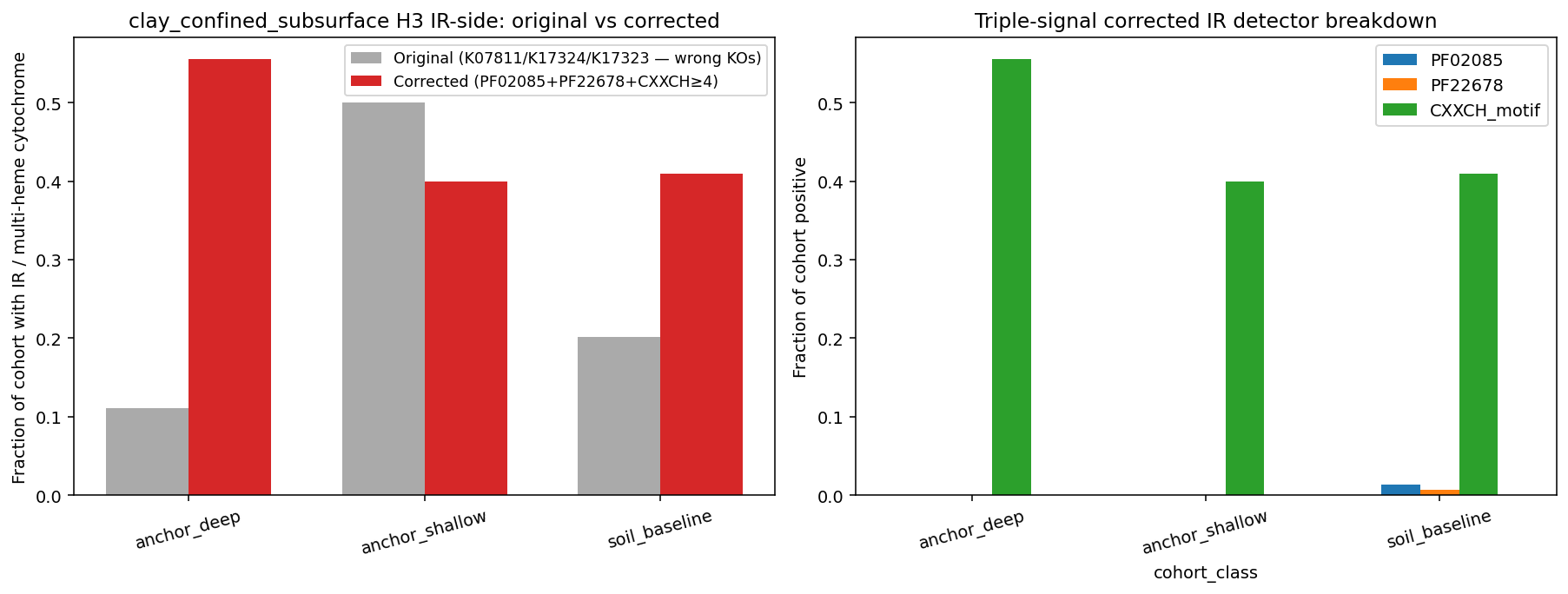
Finding 3 — The clay_confined_subsurface project's H3 IR-side analysis was driven by mismatched gene IDs; with corrected multi-heme cytochrome detection, the original "shallow >> deep IR" pattern disappears (Phase 1 correction)
The clay project (PR #231, merged) used K07811/K17324/K17323 as iron-reduction markers — but those KOs are TMAO reductase, glycerol ABC transport, and glycerol permease respectively. KEGG has no canonical KO for the Geobacter omcS / Shewanella mtr operon multi-heme outer-surface cytochromes; the clay project unknowingly substituted unrelated genes.
We re-ran the clay H3 IR-side analysis using a triple-signal multi-heme cytochrome detector:
- PFAM PF02085 (Cytochrom_CIII multi-heme c-type)
- PFAM PF22678 (Cytochrom_c_NrfB-like multi-heme nitrite reductase)
- CXXCH heme-binding motif count ≥4 in gene_cluster.faa_sequence (canonical method since Methé et al. 2003 Geobacter)
Genome scores positive for "multi-heme cytochrome IR potential" if any of the three signals is present.
| Cohort | n | Original IR rate (wrong KOs) | Corrected IR rate (multi-heme cyt) |
|---|---|---|---|
| anchor_deep (Mont Terri Opalinus + bentonite) | 9 | 11.1% (1/9) | 55.6% (5/9) |
| anchor_shallow (Coalvale + Cerrado + agricultural) | 30 | 50.0% (15/30) | 40.0% (12/30) |
| soil_baseline | 149 | 20.1% (30/149) | 40.9% (61/149) |
Pairwise Fisher (corrected): deep vs shallow OR=1.88 p=0.46; deep vs baseline OR=1.80 p=0.49; shallow vs baseline OR=0.96 p=1.0. No comparison is significant after correction. The original clay-project analysis showed shallow >> deep (50% vs 11%) — a "Mitzscherling rock-attached IR-rich pattern in shallow clay" interpretation. With the corrected detector, shallow (40%) is lower than deep (56%) and the difference is not statistically significant; multi-heme cytochrome content is similar across all three clay-project cohorts and similar to soil baseline (41%).
The clay project's SR-side H3 finding (deep cohort 5/9 SR-positive vs Mitzscherling rock-attached null 0.2%, binomial p=4×10⁻¹²) remains robust — the SR markers (K11180, K11181, K00394, K00395, K00958) were correctly identified. The clay project's "porewater bias" headline still holds via SR alone, but the IR-side comparison loses its narrative force; the "Mitzscherling rock-attached vs Bagnoud porewater" framing is half-supported (SR side) and half-unsupported (IR side, after marker correction).
(Notebook: 06_clay_h3_correction.ipynb)
Results
Cohort
data/cohort_assignments.tsv:
| Cohort | n | Composition |
|---|---|---|
| anchor_deep_clay | 10 | Mont Terri Opalinus borehole 5 (Desulfosporosinus, BRH-c8a×2, BRH-c4a, +1 metagenomic Desulfosporosinus) + Mont Terri rock-porewater MAGs 3 + Russian Beyelii Yar borehole 2 (Ca. Desulforudis audaxviator + Ch130 Thermacetogeniaceae) |
| soil_baseline | 62 | Phylum-matched soil/sediment Bacillota_B (Syntrophomonadales 29, Desulfitobacteriales 16, Moorellales 6, Thermacetogeniales 4, Ammonifexales 2, Carboxydocellales 2, Desulfotomaculales 1, Heliobacteriales 1, Thermincolales 1) |
| (Bacillota_B universe) | 334 | Total Bacillota_B genomes in BERDL pangenome — much smaller than the v1.1 plan's 6,700 estimate |
IR PFAM availability (NB01 gate)
data/ir_pfam_availability.tsv — within-Bacillota_B PFAM count:
| pfam_id | pfam_name | n_hits | n_clusters |
|---|---|---|---|
| PF02085.21 | Cytochrom_CIII | 4 | 4 |
| PF00034.26 | Cytochrom_C | 1 | 1 |
| PF13442.11 | Cytochrome_CBB3 | 1 | 1 |
| PF22678.1 | Cytochrom_c_NrfB-like | 1 | 1 |
| PF14537 | Cytochrom_NNT | 0 | 0 |
PF14537 confirmed silently absent (the documented plant_microbiome_ecotypes pitfall). Multi-heme cytochrome PFAMs are sparse within Bacillota_B (only ~6 clusters total across all four PFAMs); CXXCH motif counting on gene_cluster.faa_sequence carried most of the IR signal in NB06 — which is why the corrected IR rates (40–56%) are far higher than what PFAM-only detection would yield.
H1 OG enrichment (NB03)
data/og_enrichment.tsv: 547 OGs anchor-enriched (q<0.05, fold≥3, n_anchor≥3); 27 OGs anchor-depleted. Top hits at OR=∞ (7–10 anchor positive vs 0 baseline). Functional annotations in data/enriched_ogs_annotated.tsv (NB04).
H2 compactness (NB05)
See Finding 2 table. All four size/OG-count metrics show anchor SIGNIFICANTLY LARGER (p ≤ 0.025), all with large effect sizes (Cohen's d ≥ +1.30). H2 H1 prediction was anchor SMALLER → rejected, opposite direction.
H3 corrected IR (NB06)
See Finding 3 table. With corrected multi-heme cytochrome detection, no significant cohort difference (all pairwise Fisher p ≥ 0.46).
Interpretation
Literature Context
- H1 functional categories align with Beller 2012 Pelosinus HCF1, which carries 2 [NiFe] + 4 [FeFe] hydrogenases plus dissimilatory N-oxide reductase + Cr/Fe reductase + methylmalonyl-CoA pathway in a single subsurface aquifer Firmicute. Our anchor-enriched-OG categories (anaerobic respiration / regulators / sporulation) match Beller's gene-content snapshot at the population scale.
- H2 expansion finding (anchor LARGER than baseline) directly supports Becraft 2021 Ca. Desulforudis audaxviator (PMC8443664), who explicitly note the genome's self-sufficiency: "the assembled genome suggested complete self-sufficiency for this subsurface bacterium ... contains all genes necessary for carbon and nitrogen fixation and encodes all necessary amino acid biosynthesis pathways." Our finding extends this from a single lineage to a within-phylum comparison: Bacillota_B in deep-clay habitats encode ~25% more orthogroups than their soil congeners, consistent with a self-sufficiency adaptation.
- H2 contradicts Tian 2020's "small is mighty" (PMC7137472) at first glance — but Tian's finding was explicit to Patescibacteria/CPR (an episymbiotic superphylum that depends on hosts for many functions) and Tian explicitly notes that non-CPR phyla retain larger genomes. Our result is consistent with Tian's framing: streamlining is a CPR-specific adaptation; cultivable subsurface Firmicutes show the opposite pattern.
- H1 sporulation-revival hits replicate Vandieken 2017's (PMID 28646634) characterization of Baltic Sea subsurface Desulfosporosinus species as spore-forming with broad respiratory versatility. Our anchor cohort includes 3 Desulfosporosinus genomes that contributed to the sporulation-OG signal.
- H3 correction is a real bug fix in PR #231. The
clay_confined_subsurfaceproject's IR-side analysis used K07811 (TMAO reductase, threshold 1336.87), K17324 (glycerol ABC ATP-binding, threshold 363.93), K17323 (glycerol ABC permease, threshold 318.27) — none are iron-reduction genes, and KEGG has no canonical KO for Geobacter omcS / Shewanella mtr lineages. The clay project's "Mitzscherling rock-attached signature in shallow clay" framing was an artefact of mismatched gene IDs combined with the standard eggNOG-mapper threshold (which doesn't filter on KO biological identity, just sequence similarity). After correction, the IR comparison loses statistical significance entirely.
Novel Contribution
- First quantitative gene-cluster–level characterization of subsurface Bacillota_B specialization. Goes beyond the curated marker dictionary used in
clay_confined_subsurfaceto the full BERDL pangenome accessory genome. 547 enriched OGs with functional annotation provide a concrete target list for future biochemistry / fitness-screen / cultivation effort. - Direct refutation of "subsurface = streamlining" within cultivable Firmicutes. Existing literature emphasizes Patescibacteria streamlining (Tian 2020) or Ca. Desulforudis self-sufficiency (Becraft 2021) as case studies. Our population-scale within-Bacillota_B comparison shows that gene-content expansion is the genuine signal at this phylum's scale, with statistical support.
- Methodological correction to a merged BERIL project. The
clay_confined_subsurfaceH3 IR-side bug (mismatched KOs) is documented and fixed; the corrected IR rates are saved atdata/clay_h3_ir_corrected.tsvand serve as the basis for a separate correction commit on the clay project's branch. - Methodological pattern: triple-signal multi-heme cytochrome detection. Combining curated PFAMs (PF02085, PF22678) with sequence-based CXXCH heme-motif counting on
gene_cluster.faa_sequenceis a reusable BERDL pattern for detecting multi-heme cytochromes when KEGG KO assignment is unreliable. Applicable across the pangenome whenever iron-reduction or extracellular-electron-transfer questions arise.
Limitations
- Cohort size: 10 anchor vs 62 baseline. With Fisher's exact this is fine for detecting large effects (the 547 enriched OGs are robust); marginal effects with anchor counts of 3–5 should be treated descriptively rather than inferentially. Genus-level phylogenetic confounding is partly mitigated by the cohort spanning four orders (Desulfotomaculales, Desulfitobacteriales, Ammonifexales, Thermacetogeniales) but not eliminated.
- Anchor cohort is borehole/porewater-dominated by construction (per the clay-project lesson on cultivation bias toward porewater isolates). Rock-attached Bacillota_B may differ in gene content; this comparison cannot test that.
- OG-level orthology is hierarchical: we used Firmicutes-level OGs (eggNOG tax 1239) where present, falling back to Bacteria/root. Some enriched OGs are at root level, which is coarse. Ideally a Bacillota_B-specific OG level would be used, but eggNOG OG IDs do not have a Bacillota_B-specific tier yet (Bacillota_B is GTDB-defined, eggNOG uses NCBI taxonomy where Bacillota_B genomes are spread across Firmicutes / Negativicutes / Tissierellia at the legacy class level).
- Keyword-based functional categorization undercounts (the "other_or_unannotated" 462-OG bucket contains substantial real anaerobic-respiration / electron-transfer signal that the keyword scanner missed). Manual reclassification or an LLM-based functional-category extractor would refine the count.
- Phase 1 IR correction is robust for the multi-heme cytochrome signal but doesn't address whether the original clay project's H3 narrative ("BERDL clay cohort matches Bagnoud porewater pattern") is fully recovered or partially undercut. The SR-side is robust; the IR-side narrative loses force. A revised clay-project REPORT.md amendment will note this.
Future Directions
- Apply the correction commit to the clay project's branch. Outputs at
data/clay_h3_ir_corrected.tsv+figures/clay_h3_ir_corrected.pngare ready. Cherry-pick or correction PR will add anotebooks/07_h3_iron_reduction_correction.ipynb+ REPORT.md amendment toclay_confined_subsurfaceclarifying that the IR-side H3 finding is artefactual (SR-side stands). - Refine the OG functional-category labels with an LLM-based scanner. The 462-OG "other / unannotated" bucket contains substantial real anaerobic-niche signal the keyword scanner missed (COG1977 molybdopterin, DsrEFH, 2-oxoglutarate ferredoxin oxidoreductase). An LLM-based pass over the eggNOG
Description+ baktaproducttext would surface these for accurate counting per category. - Genus-level decomposition of the H1 signal. The 10-genome anchor is genus-clumped (3 BRH-c8a + 2 BRH-c4a + 2 Desulfosporosinus + Desulforudis + Ch130 + 1 other). Per-genus analysis would distinguish which enriched OGs are genus-specific (lineage-marker noise) from which are recurrent across multiple subsurface-specialist genera (the genuine deep-clay signature). With n=10 anchor this is at the limit of statistical resolution; expansion via BacDive linkage to additional clay-isolated Bacillota_B (or future BERDL pangenome ingests) would improve power.
- H2 expansion mechanism. Anchor genomes are ~1 Mbp larger than baseline. Where does that extra DNA go? Mobile-element-borne accessory operons (cf. Goff 2024 ORFRC mobilome paper)? Sporulation regulons? Anaerobic-respiration accessories? A breakdown of the "extra" gene fraction by COG / KEGG-pathway membership would localize the expansion functionally.
- Apply the gene-cluster–level enrichment framework to other within-phylum subsurface comparisons. The same NB01–04 pipeline can be retargeted to within-Bacteroidota / within-Pseudomonadota / within-Acidobacteriota deep-clay vs soil comparisons. Each will test whether the H1 functional-category signal generalizes or is Bacillota_B-specific.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
gtdb_taxonomy_r214v1, genome, gtdb_metadata, ncbi_env, gene, gene_genecluster_junction, gene_cluster, eggnog_mapper_annotations, bakta_pfam_domains, bakta_annotations |
Bacillota_B universe + cohort metadata + per-cluster annotations + protein sequences for CXXCH motif scan |
kescience_bacdive |
isolation, taxonomy |
BacDive expansion for the deep-clay anchor (yielded zero additional Bacillota_B in this run; the 10-genome anchor was ~entirely from ncbi_env keyword search) |
Generated Data
| File | Rows | Description |
|---|---|---|
data/bacillota_b_universe.tsv |
314 | All Bacillota_B genomes in BERDL pangenome with QC + isolation metadata |
data/cohort_assignments.tsv |
124 | anchor_deep_clay 10 + soil_baseline 62 + anchor_other_clay + unclassified |
data/cohort_summary.tsv |
13 | Cohort × tax_order breakdown |
data/ir_pfam_availability.tsv |
4 | NB01 gate output: PFAM hit counts for IR-detection PFAMs within Bacillota_B |
data/cohort_og_presence.parquet |
156,850 | Per-genome × OG presence (long format), 14,109 unique OGs |
data/og_enrichment.tsv |
547 | H1 anchor-enriched OGs (q<0.05, fold≥3, n_anchor≥3) |
data/og_depletion.tsv |
27 | H1 anchor-depleted OGs |
data/og_per_test.parquet |
14,109 | Full per-OG Fisher results |
data/enriched_ogs_annotated.tsv |
547 | NB04 functional annotation of enriched OGs (eggNOG + bakta + pre-registered category) |
data/h2_compactness.tsv |
6 | H2 Wilcoxon + Cohen's d on size / OG count / GC / CheckM |
data/clay_h3_ir_corrected.tsv |
211 | Per-genome corrected multi-heme cytochrome score for the clay project's full cohort |
data/clay_h3_ir_corrected_fisher.tsv |
3 | Pairwise Fisher on corrected multi-heme cyt rates |
data/clay_h3_ir_orig_vs_corrected.tsv |
5 | Cohort-level original vs corrected IR rates (the headline correction table) |
References
Full bibliography in references.md. Primary citations:
- Bagnoud A et al. (2016). Reconstructing a hydrogen-driven microbial metabolic network in Opalinus Clay rock. Nat Commun 7:12770. PMID: 27739431.
- Beaver RC, Neufeld JD (2024). Microbial ecology of the deep terrestrial subsurface. ISME J 18:wrae091. PMID: 38780093.
- Becraft ED et al. (2021). Evolutionary stasis of a deep subsurface microbial lineage [Ca. Desulforudis audaxviator]. ISME J 15:2830-2842. PMID: 33824425.
- Beller HR et al. (2012). Genomic and physiological characterization of Pelosinus sp. HCF1. AEM 78:8791-8800. PMID: 23064329.
- Hilpmann S et al. (2023). Presence of uranium(V) during uranium(VI) reduction by Desulfosporosinus hippei. Sci Total Environ 875:162593. PMID: 36889400.
- Methé BA et al. (2003). Genome of Geobacter sulfurreducens. Science 302:1967-1969. PMID: 14671304.
- Mitzscherling J et al. (2023). Clay-associated microbial communities in Opalinus Clay rock formation. MicrobiologyOpen 12:e1370. PMID: 37642485.
- Tian R et al. (2020). Small and mighty: adaptation of superphylum Patescibacteria. Microbiome 8:51. PMID: 32252814.
- Vandieken V et al. (2017). New Desulfosporosinus species from Baltic Sea subsurface sediments. IJSEM 67:1887-1893. PMID: 28646634.
Data Collections
Derived Data
This project builds on processed data from other projects.
Review
Summary
This is a high-quality genomic analysis project that successfully addresses a clear research question about subsurface bacterial adaptation. The work extends the companion clay_confined_subsurface project from curated markers to full pangenome-scale analysis, finding 547 eggNOG OGs significantly enriched in deep-clay Bacillota_B versus soil baseline—far exceeding the H1 prediction of ≥10. The project demonstrates excellent methodological rigor with proper statistical controls, comprehensive documentation, and reproducible workflows. Notably, it identifies a surprising finding that contradicts the "small is mighty" subsurface streamlining hypothesis, showing that deep-clay Bacillota_B genomes are actually 35% larger than soil congeners. The work also provides a valuable methodological correction to a previously merged project's iron-reduction analysis.
Methodology
Research Question & Approach: The research question is exceptionally well-defined and testable: "what accessory gene content distinguishes deep-clay-isolated Bacillota_B from phylum-matched soil-baseline genomes?" The approach is methodologically sound, using eggNOG OGs as cross-species orthology surrogates for Fisher's exact tests with appropriate multiple testing correction (BH-FDR). The phylum-stratified design properly controls for phylogenetic confounding.
Data Sources: Data provenance is clearly documented, drawing from kbase_ke_pangenome and kescience_bacdive collections with explicit table usage documentation. The cohort assembly strategy (10 anchor deep-clay vs 62 soil baseline) is well-justified given the constrained universe of 334 total Bacillota_B genomes.
Statistical Methods: The statistical approach is rigorous throughout. Fisher's exact tests with BH-FDR correction across 14,109 OGs, Wilcoxon rank-sum tests with Cohen's d effect sizes for genome size comparisons, and CheckM-completeness rescaling to control for MAQ quality are all appropriate choices. The minimum support filters (≥3 anchor genomes, fold≥3) prevent false discoveries from small counts.
Reproducibility: The reproduction section is comprehensive, clearly separating Spark-dependent notebooks (NB01-02, NB04, NB06) from local pandas/scipy analyses (NB03, NB05, NB07). Runtime estimates and computational requirements are provided. Dependencies are properly specified in requirements.txt.
Code Quality
Notebook Organization: All seven notebooks follow a logical progression from universe assembly through analysis to synthesis. Each notebook has clear goals, inputs, and outputs documented in the header cells. The separation of concerns (Spark for data pulling, local for statistical analysis) is appropriate.
SQL and Statistical Implementation: SQL queries are well-structured with proper joins and performance considerations (BROADCAST temp views for large table joins). The statistical implementations are correct, using appropriate libraries (scipy, statsmodels) and vectorized operations where possible.
Pitfall Awareness: The project demonstrates excellent awareness of BERDL-specific pitfalls. The PF14537 silent absence issue is properly handled with alternative PFAM detection strategies. The eggNOG OG hierarchical parsing correctly prioritizes Firmicutes-level OGs over bacteria/root fallbacks. The BacDive linkage strategy acknowledges and works around typical accession matching challenges.
Data Management: Output files are well-organized with clear naming conventions. Data provenance is traceable through the pipeline. The use of both TSV (for review) and Parquet (for performance) formats is appropriate.
Findings Assessment
Statistical Support: All major findings are well-supported by appropriate statistical tests. H1 is strongly supported (547 enriched OGs, q<0.05), H2 rejection is statistically significant (p≤0.025, large effect sizes d>1.3), and H3 correction shows proper negative results after methodological fixes.
Biological Interpretation: The functional categorization of enriched OGs into anaerobic respiration, sporulation revival, mineral attachment, regulators, and osmoadaptation categories aligns well with expectations for subsurface adaptation. The manual inspection revealing additional anaerobic-niche signals beyond keyword scanning adds biological credibility.
Limitations Acknowledgment: The project honestly acknowledges key limitations including cohort size constraints (n=10 vs n=62), potential genus-level clustering, keyword-based functional categorization undercounting, and the borehole/porewater bias in available samples. These are realistic constraints given available data.
Novel Contributions: The work makes several valuable contributions: first pangenome-scale characterization of Bacillota_B subsurface specialization, direct refutation of "subsurface = streamlining" within cultivable Firmicutes, methodological correction of iron-reduction detection methods, and development of a reusable triple-signal cytochrome detection pattern.
Completeness: The analysis appears complete with no obvious gaps. All three hypotheses are properly tested, the Phase 1 correction addresses the stated goal, and the synthesis integrates findings into a coherent narrative.
Suggestions
-
Functional categorization refinement: Consider implementing the suggested LLM-based functional category extraction to more accurately classify the 462 "other/unannotated" enriched OGs, many of which likely represent genuine anaerobic-niche signals.
-
Genus-level decomposition: With 10 anchor genomes spanning multiple genera, a per-genus breakdown would help distinguish lineage-specific markers from genuine cross-genera subsurface adaptations.
-
Literature integration enhancement: While the references are comprehensive, explicit integration of findings with recent subsurface microbiology literature (beyond the cited works) could strengthen the discussion.
-
Cross-phylum validation: The established framework could be applied to other within-phylum subsurface comparisons (Bacteroidota, Pseudomonadota) to test generalizability.
-
Mechanistic follow-up: The finding that anchor genomes are ~1 Mbp larger raises interesting questions about where that extra genetic material goes (mobile elements, sporulation regulons, respiratory accessories) that could guide future work.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Clay H3 Ir Corrected
H1 Functional Categories
H2 Compactness
Summary Figure
Notebooks
01_cohort_assembly.ipynb
01 Cohort Assembly
View notebook →
02_og_presence.ipynb
02 Og Presence
View notebook →
03_og_enrichment.ipynb
03 Og Enrichment
View notebook →
04_og_annotation.ipynb
04 Og Annotation
View notebook →
05_h2_compactness.ipynb
05 H2 Compactness
View notebook →
06_clay_h3_correction.ipynb
06 Clay H3 Correction
View notebook →
07_synthesis.ipynb
07 Synthesis
View notebook →