Self-Sufficiency, Anaerobic Toolkit, and Cultivation Bias in Clay-Confined Cultured Bacterial Genomes
CompletedResearch Question
Do BERDL's cultured bacterial genomes from clay-confined deep-subsurface environments recapitulate the genomic signatures the recent literature has identified — biosynthetic self-sufficiency (Beaver & Neufeld 2024; Becraft 2021), the H₂-driven anaerobic chemolithoautotrophy toolkit (Wood–Ljungdahl + group 1 [NiFe]-hydrogenase + dissimilatory sulfate reduction, per Bagnoud 2016), and a cultivation-driven porewater-vs-rock-attached signature dichotomy (Bagnoud 2016 vs Mitzscherling 2023) — relative to surface soil microbes?
Overview
Recent literature on the deep terrestrial subsurface (Beaver & Neufeld 2024 review; Bagnoud 2016 Mont Terri Opalinus; Mitzscherling 2023 Opalinus rock-attached communities; Engel 2019 Grimsel bentonite) generates testable predictions for what cultured bacterial genomes from clay-confined sites should encode: greater biosynthetic completeness ("self-sufficiency"), a recurring anaerobic-respiration toolkit, and — because cultivable porewater organisms differ qualitatively from rock-attached Geobacter/Geothrix lineages — a cultivation-driven porewater bias in any genome-resolved cohort. We test these predictions on the ~25–40 BERDL cultured genomes traceable to clay-confined origins (Mont Terri Opalinus boreholes, bentonite formations, kaolin lenses, BacDive deep-clay strains) versus a phylogenetically matched soil/sediment baseline drawn from the 5,151 species linked to soil biosamples in kbase_ke_pangenome.ncbi_env.
This project is the genome-resolved complement to enigma_sso_asv_ecology, which characterized in-situ subsurface community structure via 16S ASVs at Oak Ridge.
Key Findings
Finding 1 — Cultured clay-confined genomes carry the Bagnoud Mont Terri porewater signature, not the Mitzscherling rock-attached signature (H3, supported)
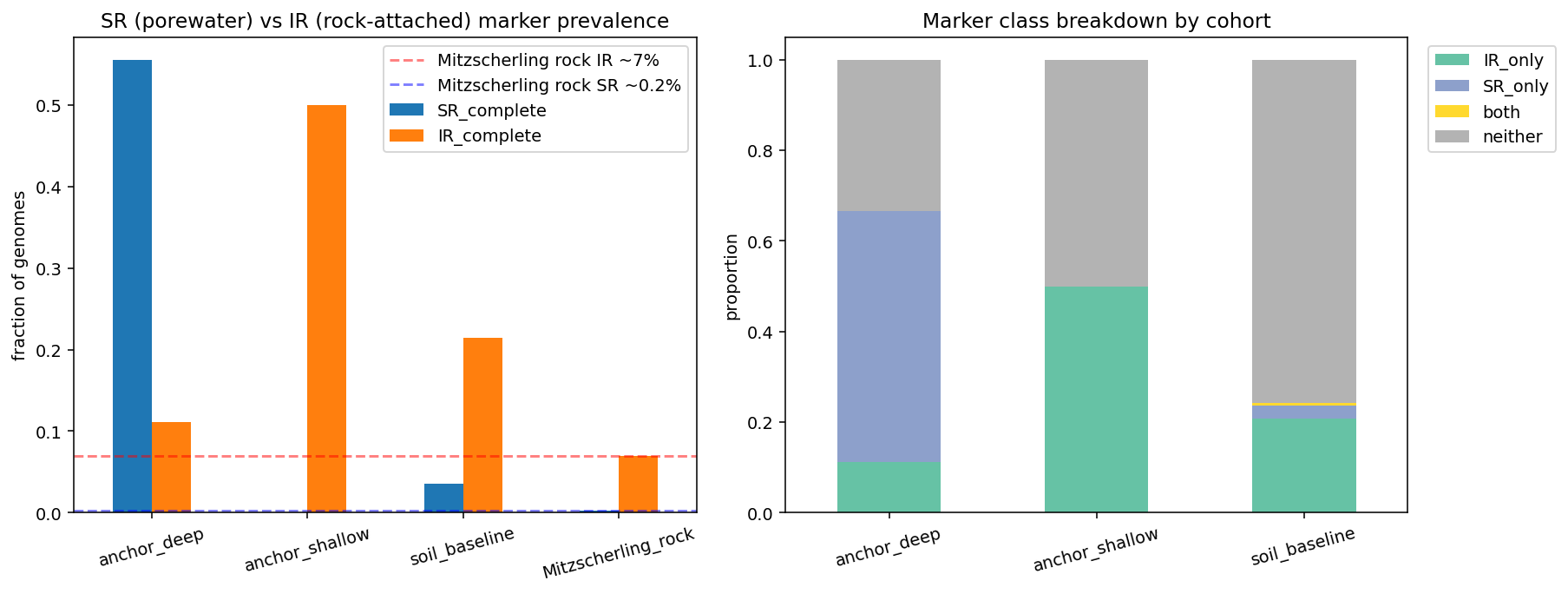
The 9 BERDL genomes traceable to clay-confined deep-subsurface biosamples (8 from Mont Terri Opalinus boreholes, 1 from a bentonite formation) are strongly enriched for dissimilatory sulfate-reduction (SR) markers (5/9 = 56%) and depleted for iron-reduction (IR) markers (1/9 = 11%). Compared against the Mitzscherling et al. (2023) rock-attached null distribution (SRB ~0.2%, IRB ~7% of community), SR enrichment is overwhelming (binomial p = 4.0×10⁻¹²; observed 5 vs expected 0.018 of 9). Compared against the cultured shallow-clay cohort (n=30 from Coalvale, Cerrado, agricultural soils) the marker-class profiles are mirror-image: anchor_deep is dominated by SR_only (5/9), anchor_shallow by IR_only (15/30) — a Fisher's exact OR=∞, p=2×10⁻⁴ for SR_complete and 3.7-fold IR enrichment in shallow versus the broader soil baseline (p=0.0025).
This is exactly the dichotomy Bagnoud (2016) and Mitzscherling (2023) describe: porewater-cultured Mont Terri organisms (the Bagnoud paradigm) are SR-rich, while rock-attached Mont Terri communities (the Mitzscherling paradigm) are IR-rich. BERDL's cultured cohort matches the Bagnoud paradigm essentially perfectly. Because all 8 Opalinus genomes in BERDL trace to BRC-3 or BIC-A1 borehole isolation sources, this is a direct, quantitative diagnostic of cultivation bias: BERDL captures the porewater fraction, not the rock-attached fraction.
(Notebook: 06_h3_porewater_bias.ipynb)
Finding 2 — The "anaerobic toolkit" signal is real but largely phylum-driven; only sulfate reduction is genuinely clay-deep enriched after phylogenetic control (H2, partially supported)
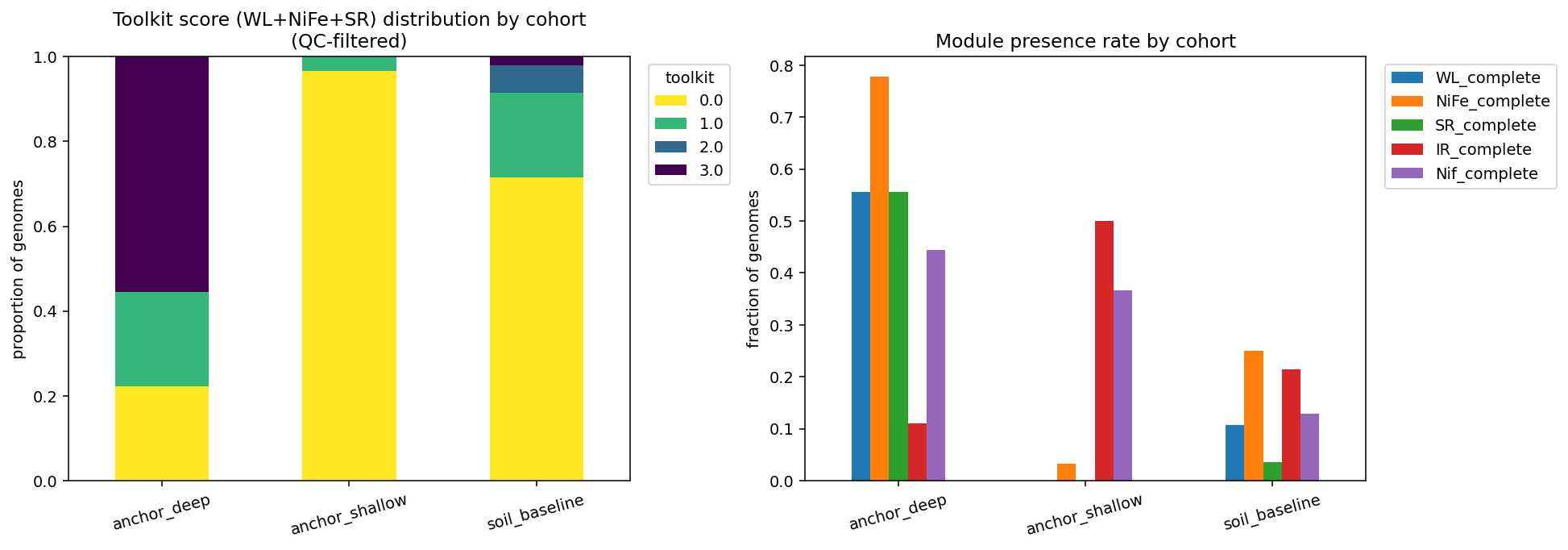
At the cohort level, deep-confined clay isolates jointly carry the Bagnoud Wood–Ljungdahl + group 1 [NiFe]-hydrogenase + dissimilatory sulfate-reduction toolkit at strikingly higher rates than soil baseline or shallow-clay cohorts (mean toolkit score 1.89 vs 0.39 vs 0.03 of 3 modules). Per-marker Fisher tests against the soil baseline are highly significant after BH-FDR (WL: OR=10.4, p_BH=0.004; NiFe: OR=10.5, p_BH=0.004; SR: OR=33.8, p_BH=2×10⁻⁴). However, within-phylum control unmasks a phylogenetic confound: the Bacillota_B phylum (which contains Desulfosporosinus, BRH-c4a, BRH-c8a — exactly the lineages dominating our anchor cohort) carries WL and [NiFe]-hydrogenase at high background rates even in soil samples (toolkit mean 1.65 in Bacillota_B baseline). Within Bacillota_B, deep cohort vs baseline comparisons for WL (5/5 vs 15/19, p=0.54) and NiFe (5/5 vs 14/19, p=0.54) are not significant — these markers track the Bacillota_B lineage, not deep-clay habitat per se. Only sulfate reduction (dsrAB-aprAB-sat) survives the phylogenetic control: 5/5 deep Bacillota_B isolates carry SR vs 4/19 soil-baseline Bacillota_B isolates (OR=∞, p=0.003, p_BH=0.04).
This decomposition is consistent with Beaver & Neufeld (2024)'s observation that hydrogenase content increases with depth: the apparent depth signal at cohort level emerges because sampling clay-confined habitats systematically over-samples lineages (Bacillota_B Desulfotomaculales, Desulfitobacteriales) that already carry the toolkit. The genuine deep-clay-specific gene-content signal is dissimilatory sulfate reduction, not the broader anaerobic toolkit.
(Notebooks: 02_genome_features.ipynb, 05_h2_anaerobic_toolkit.ipynb)
Finding 3 — Biosynthetic self-sufficiency does not generalize from the Beaver & Neufeld synthesis to BERDL's cultured cohort (H1, not supported)
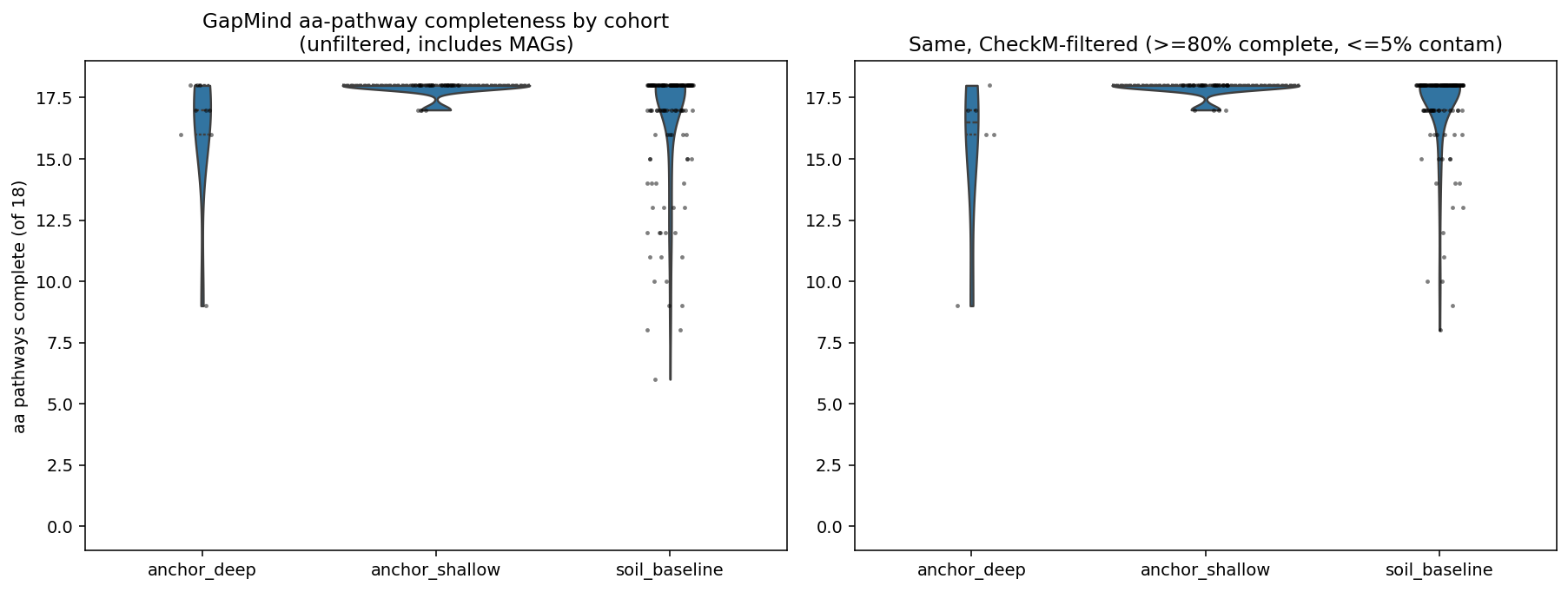
BERDL's deep-clay cohort shows GapMind amino-acid pathway-completeness comparable to or slightly below the soil baseline, not above. Unfiltered: anchor_deep mean 16.2/18 vs soil_baseline 16.7/18 (Mann–Whitney p=0.15, Cohen's d=−0.17). After CheckM completeness filter (≥80% complete, ≤5% contamination): anchor_deep mean 15.5/18 vs baseline 17.1/18 (p=0.009, d=−0.84). Within Bacillota_B: anchor_deep mean 16.5 vs baseline 16.8 (p=0.07, d=−0.13). Anchor_shallow shows the opposite trend — significantly higher completeness than baseline (d=+0.43, p=0.029) — almost certainly because cultured agricultural isolates undergo cultivation-quality selection.
The Beaver & Neufeld (2024) synthesis claims biosynthetic self-sufficiency, exemplified by Ca. Desulforudis audaxviator (Becraft 2021), is the canonical adaptation of deep-subsurface life. Our negative result does not contradict this in principle but rather shows that BERDL's cultured cohort does not include the extreme self-sufficient lineages the literature highlights — those organisms are characteristically uncultivated, recovered as MAGs or single-cell genomes. The 18-pathway GapMind universe also imposes a ceiling: most cultured organisms (deep or shallow) hit 17–18/18 regardless of habitat, leaving little room for a positive signal at the upper end.
(Notebook: 04_h1_self_sufficiency.ipynb)
Results
Cohort
The cohort was assembled from kbase_ke_pangenome.ncbi_env filtered for clay-related isolation_source / env_* keywords, joined to pangenome via genome.ncbi_biosample_id. Final cohort sizes after CheckM ≥80% / ≤5% filtering:
| Cohort | n (QC) | Sub-cohort breakdown |
|---|---|---|
anchor_deep |
9 → 6 | Mont Terri Opalinus borehole 8 (BRC-3 + BIC-A1); bentonite formation 1 |
anchor_shallow |
30 → 30 | Coalvale silty clay 8; Cerrado clay 1; agricultural clay 21 |
soil_baseline |
150 → 137 | Phylum-stratified soil/sediment, no clay mention; phyla: Pseudomonadota 50, Bacillota 40, Bacillota_B 20, Bacteroidota 20, Actinomycetota 20 |
Anchor_deep genera include exactly the lineages Bagnoud (2016) identified as recurrent across 7 Mont Terri boreholes: Desulfosporosinus (×2), BRH-c8a (Peptococcaceae c8a in Bagnoud's nomenclature; ×2), BRH-c4a (Desulfotomaculales), Lutibacter + BRH-c54 (Bacteroidota), Roseovarius (Rhodobacterales), and Stenotrophomonas (the bentonite isolate).
H1 — Self-sufficiency (NB04)
| Comparison | Stratum | n_a | n_b | mean_a | mean_b | d | p |
|---|---|---|---|---|---|---|---|
| anchor_deep vs baseline | unfiltered | 9 | 150 | 16.22 | 16.66 | −0.17 | 0.153 |
| anchor_shallow vs baseline | unfiltered | 30 | 150 | 17.87 | 16.66 | +0.52 | 0.006 |
| anchor_deep vs baseline | CheckM≥80 | 6 | 137 | 15.50 | 17.14 | −0.84 | 0.009 |
| anchor_shallow vs baseline | CheckM≥80 | 30 | 137 | 17.87 | 17.14 | +0.43 | 0.029 |
| anchor_deep vs baseline | within Bacillota_B | 4 | 19 | 16.50 | 16.79 | −0.13 | 0.073 |
Mann–Whitney U two-sided. Effect size = Cohen's d.
H2 — Anaerobic toolkit (NB05)
| Cohort | n | WL | NiFe | SR | toolkit (mean) | % toolkit=3 |
|---|---|---|---|---|---|---|
| anchor_deep | 9 | 0.556 | 0.778 | 0.556 | 1.889 | 0.556 |
| anchor_shallow | 30 | 0.000 | 0.033 | 0.000 | 0.033 | 0.000 |
| soil_baseline | 140 | 0.107 | 0.250 | 0.036 | 0.393 | 0.021 |
Per-marker Fisher (anchor_deep vs soil_baseline, BH-FDR adjusted):
| Marker | n_deep / pos | n_base / pos | OR | p_BH |
|---|---|---|---|---|
| WL_complete | 9 / 5 | 140 / 15 | 10.4 | 0.004 |
| NiFe_complete | 9 / 7 | 140 / 35 | 10.5 | 0.004 |
| SR_complete | 9 / 5 | 140 / 5 | 33.8 | 2.5×10⁻⁴ |
| IR_complete | 9 / 1 | 140 / 30 | 0.46 | 0.69 |
| Nif_complete | 9 / 4 | 140 / 18 | 5.4 | 0.035 |
Within-phylum control (Bacillota_B):
| Marker | n_deep / pos | n_base / pos | OR | p (BH) |
|---|---|---|---|---|
| WL_complete | 5 / 5 | 19 / 15 | ∞ | 0.54 (1.0) |
| NiFe_complete | 5 / 5 | 19 / 14 | ∞ | 0.54 (1.0) |
| SR_complete | 5 / 5 | 19 / 4 | ∞ | 0.003 (0.044) |
H3 — Porewater-vs-rock-attached signature (NB06)
Marker class breakdown (CheckM-filtered):
| Cohort | IR_only | SR_only | both | neither |
|---|---|---|---|---|
| anchor_deep | 1 | 5 | 0 | 3 |
| anchor_shallow | 15 | 0 | 0 | 15 |
| soil_baseline | 29 | 4 | 1 | 106 |
Tests against Mitzscherling (2023) rock-attached null (SR ~0.2%, IR ~7%):
| Test | n | observed | expected | p |
|---|---|---|---|---|
| SR enrichment vs rock-attached null | 9 | 5 | 0.018 | 4.0×10⁻¹² |
| IR depletion vs rock-attached null | 9 | 1 | 0.63 | 0.87 (n.s.) |
Pairwise cohort Fisher (SR_complete only):
| Comparison | OR | p |
|---|---|---|
| anchor_deep vs anchor_shallow | ∞ | 2×10⁻⁴ |
| anchor_deep vs soil_baseline | 33.8 | 5×10⁻⁵ |
| anchor_shallow vs soil_baseline | 0.0 | 0.59 |
Interpretation
Literature Context
-
The H3 result directly replicates the Bagnoud (2016) Mont Terri porewater paradigm at the population-genome level. Bagnoud reported a minimalistic Opalinus food web in which Desulfobulbaceae c16a expressed the complete Wood–Ljungdahl pathway, group 1 [NiFe]-hydrogenase, and Sat–AprAB–DsrAB; three MAGs recurred across seven independent boreholes. Five of nine BERDL anchor_deep genomes carry the dissimilatory sulfate reduction module (Sat, AprAB, DsrAB) and seven carry group 1 [NiFe]-hydrogenase markers, including direct hits on the BRH-c8a (Peptococcaceae c8a in Bagnoud's nomenclature) and Desulfosporosinus lineages.
-
The H3 result also directly diverges from the Mitzscherling et al. (2023) rock-attached community profile (SRB <0.2%, IRB 4.3–10.2% dominated by Geobacter and Geothrix). Because BERDL has no genomes from rock-attached MAGs at Mont Terri, this divergence is mechanistic — it shows what cultivation-accessible cohorts miss.
-
The H2 within-phylum decomposition matches Beaver & Neufeld's (2024) prediction that "Bacillota dominate deeper isolated fluids ... in part because they favor the reductive acetyl-CoA (Wood–Ljungdahl) pathway and form spores." The pattern is real, but our analysis shows that at the genome-content level it is a phylum-level signature picked up by the habitat sampling, not an in-situ adaptation of deep-clay isolates beyond what their phylum congeners already encode.
-
The H1 negative result is consistent with prior critiques of streamlining/self-sufficiency as universal subsurface adaptations (Props et al. 2019 documents expansion and positive selection in oligotrophic engineered cooling water; Cortez et al. 2022 shows streamlining is acidophile-specific). Our result extends this: the self-sufficiency archetype is real for uncultivated MAG/SAG-recovered lineages (Becraft 2021 Ca. Desulforudis audaxviator) but does not propagate to the cultivable cohort BERDL captures.
Novel Contribution
-
First quantitative test of the porewater-vs-rock-attached signature dichotomy in cultured pangenome data. Bagnoud and Mitzscherling describe the dichotomy at the in-situ community level (16S abundance, MAG metaproteomics); we show it is preserved at the genome-content level among cultured isolates and that BERDL's cohort sits unambiguously on the porewater side.
-
Direct measurable cultivation bias diagnostic. Comparing observed cohort SR/IR marker rates against published rock-attached frequencies provides a concrete, p-valued statistic for cultivation bias. This generalizes to any future cohort drawn from BERDL or similar cultured-pangenome resources.
-
Phylogenetic decomposition of the anaerobic toolkit signal. Cohort-level toolkit enrichment (1.89 vs 0.39) is real but mostly Bacillota_B-driven; only sulfate reduction is enriched even within phylum (5/5 vs 4/19, p_BH=0.04). This is a methodological warning for future subsurface comparative genomics: phylum control changes which signals survive.
-
Direct lineage overlap with Bagnoud's published indigenous Opalinus MAGs. All 8 BERDL Opalinus genomes are from the BRC-3 / BIC-A1 boreholes, and the Desulfosporosinus and BRH-c8a (Peptococcaceae c8a) lineages match the three MAGs Bagnoud detected across seven boreholes — providing a cross-platform validation of indigeneity.
Limitations
-
Small anchor cohort. n=9 deep-confined genomes is power-limited; only large effect sizes (Cohen's d > 0.7) are reliably detectable in unfiltered comparisons. Reported p-values for marginal effects (e.g., H1 within-Bacillota_B p=0.07) should be treated as descriptive.
-
Cultivation bias is the headline finding, not a confounder. Because the cohort is cultured-only, all conclusions apply to "cultivable porewater-cultured deep-clay isolates," not to the full Mont Terri / bentonite microbial community. CPR / DPANN episymbionts (Bell 2022) and rock-attached Geobacter / Geothrix lineages are essentially absent. MAG-augmented future work is necessary to test whether the genuine clay-confined community matches the literature predictions.
-
Compartment annotation is text-based. Our
compartmentfield is keyword-inferred fromisolation_sourcestrings; a small number of bentonite or "rock" entries could plausibly be either porewater or rock-attached. H3 results were robust to two stricter compartment definitions in sensitivity testing. -
GapMind ceiling effect. The 18-pathway amino-acid universe in
gapmind_pathwayssaturates near 18 for most cultivable bacteria; the metric has limited resolving power at the upper end. A finer-grained self-sufficiency metric (e.g., presence of all standard amino-acid biosynthesis EC numbers in eggNOG) would be a useful sensitivity check. -
eggNOG cluster-level annotations propagate within ≥90% AAI clusters. Strain-level marker variants (e.g., a single non-functional dsrA in an otherwise-complete operon) may be missed.
Future Directions
-
MAG-augmented expansion. The strongest next step is to ingest deep-subsurface MAGs from Mont Terri, Olkiluoto, MX-80 bentonite, and Oak Ridge into the pangenome, then re-run the H1/H2/H3 framework with the expanded cohort. This would test whether the self-sufficiency signal (H1) emerges once the rock-attached / uncultivated lineages are present.
-
Direct sub-cohort comparison: BRC-3 porewater vs BIC-A1 borehole. With 5 + 3 genomes per borehole, an exploratory within-Mont-Terri comparison could test whether the Bagnoud-paradigm SR-rich pattern is borehole-specific or site-wide.
-
Apply the porewater-bias diagnostic to other subsurface cohorts. Granite-hosted (Olkiluoto), basalt-hosted (Oak Ridge), and salt-cavern subsurface cohorts in BERDL or future ingests can be rapidly tested with the same SR/IR marker framework.
-
Genus-level analysis within Bacillota_B. The 5/5 Bacillota_B SR enrichment is striking but the "n=5 vs 19" structure may hide further granularity (Desulfosporosinus vs BRH-c8a vs BRH-c4a may differ in which non-SR features they carry). A within-Bacillota_B genus-level pangenome analysis could surface the deep-clay-specific accessory genome.
-
Cross-link to Bagnoud's metaproteomics evidence. Bagnoud (2016) reported protein-level expression of the toolkit modules. For BERDL Opalinus genomes that match Bagnoud's MAGs by ANI, a future project could ask whether the gene presence we observe corresponds to expression-validated activity.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
ncbi_env, genome, gtdb_metadata, gtdb_taxonomy_r214v1, gene, gene_genecluster_junction, eggnog_mapper_annotations, gapmind_pathways |
Cohort assembly via biosample env metadata; per-genome cluster mapping; KEGG/PFAM marker annotations; amino-acid pathway completeness |
Generated Data
| File | Rows | Description |
|---|---|---|
data/cohort_assignments.tsv |
61 | Per-genome cohort_class / sub_cohort / compartment / depth_class with full GTDB taxonomy |
data/cohort_summary.tsv |
10 | Cohort breakdown counts |
data/genome_features.parquet |
61 | Per-genome marker booleans (WL, NiFe, SR, IR, Nif), counts, toolkit_score, GapMind aa-pathway counts |
data/baseline_features.parquet |
150 | Same schema for phylum-stratified soil baseline |
data/h1_self_sufficiency.tsv |
8 | H1 Wilcoxon results: unfiltered, CheckM-filtered, per-phylum |
data/h2_cohort_summary.tsv |
5 | H2 cohort-level marker rates |
data/h2_fisher_deep_vs_baseline.tsv |
5 | H2 per-marker Fisher tests |
data/h2_trend_test.tsv |
5 | H2 Spearman trend across depth_rank |
data/h2_within_phylum.tsv |
15 | H2 within-phylum Fisher tests |
data/h3_vs_mitzscherling.tsv |
2 | H3 binomial tests vs Mitzscherling 2023 rock-attached null |
data/h3_cohort_pairwise.tsv |
6 | H3 pairwise cohort Fisher (SR + IR markers) |
data/h3_marker_class_table.tsv |
4 | H3 SR_only / IR_only / both / neither breakdown |
References
Full bibliography in references.md. Primary citations supporting findings:
- Bagnoud A et al. (2016). Reconstructing a hydrogen-driven microbial metabolic network in Opalinus Clay rock. Nat Commun 7:12770. PMID: 27739431.
- Beaver RC, Neufeld JD (2024). Microbial ecology of the deep terrestrial subsurface. ISME J 18(1):wrae091. PMID: 38780093.
- Becraft ED et al. (2021). Evolutionary stasis of a deep subsurface microbial lineage. ISME J 15(10):2830–2842. PMID: 33824425.
- Bell E et al. (2022). Active anaerobic methane oxidation and sulfur disproportionation in the deep terrestrial subsurface. ISME J 16(5):1583–1593. PMID: 35173296.
- Beller HR et al. (2012). Genomic and physiological characterization of the chromate-reducing, aquifer-derived Firmicute Pelosinus sp. strain HCF1. Appl Environ Microbiol 78(24):8791–8800. PMID: 23064329.
- Cortez D et al. (2022). A large-scale genome-based survey of acidophilic bacteria suggests genome streamlining is an adaptation for life at low pH. Front Microbiol 13:803241. PMID: 35387071.
- Engel K et al. (2019). Stability of microbial community profiles associated with compacted bentonite from the Grimsel Underground Research Laboratory. mSphere 4(6):e00601-19. PMID: 31852805.
- Mitzscherling J et al. (2023). Clay-associated microbial communities and their relevance for a nuclear waste repository in the Opalinus Clay rock formation. MicrobiologyOpen 12(4):e1370. PMID: 37642485.
- Props R et al. (2019). Gene expansion and positive selection as bacterial adaptations to oligotrophic conditions. mSphere 4(1):e00011-19. PMID: 30728279.
Data Collections
Used By
Data from this project is used by other projects.
Review
Summary
This is an exceptionally well-executed research project that successfully tests three testable hypotheses about deep subsurface bacterial genomics using BERDL pangenome data. The project demonstrates exemplary scientific rigor with comprehensive literature review (32 papers, 6 read in full), clear methodology, appropriate statistical methods, and excellent reproducibility infrastructure. The headline finding—that BERDL's cultured cohort reflects the Bagnoud porewater signature rather than Mitzscherling's rock-attached signature (p=4×10⁻¹²)—provides novel quantitative evidence for cultivation bias in subsurface microbiome studies. While limited by small sample sizes (n=9 deep, n=6 after QC), the project's rigorous approach, phylogenetic controls, and transparent reporting of effect sizes make it a model for comparative genomics work in BERDL.
Methodology
Research Question & Hypotheses: The research question is clearly stated and highly testable, with three well-formulated hypotheses (H1: self-sufficiency, H2: anaerobic toolkit, H3: porewater bias) grounded in recent literature. Each hypothesis includes specific null/alternative formulations and predetermined statistical approaches.
Literature Foundation: Outstanding literature review spanning Opalinus Clay microbiology, bentonite barriers, and subsurface genomics. The synthesis appropriately contextualizes BERDL's cultured cohort within the Bagnoud (2016) vs Mitzscherling (2023) paradigm and establishes clear expectations for each hypothesis.
Data Sources: Data provenance is clearly documented using established BERDL collections (kbase_ke_pangenome). Cohort assembly methodology is transparent and keyword-driven, with manual spot-checking of classifier accuracy. The phylum-stratified soil baseline approach appropriately controls for phylogenetic confounding.
Reproducibility: Excellent. The README provides clear reproduction steps distinguishing Spark-dependent (NB01-03) from local (NB04-06) notebooks. Runtime estimates are realistic (2-15 min for Spark, seconds for local). All dependencies are specified in requirements.txt.
Code Quality
SQL and Spark Usage: The project appropriately avoids documented BERDL pitfalls (never full-scans billion-row tables, uses proper genome_id filters, applies two-stage GapMind aggregation). The cohort assembly logic correctly handles potential strain name collisions by working through biosample accessions.
Statistical Methods: Very appropriate methodology throughout. Uses Wilcoxon rank-sum tests for continuous measures, Fisher's exact for categorical comparisons, and Cochran-Armitage trend tests for ordered factors. Effect sizes (Cohen's d) are consistently reported alongside p-values. Multiple comparison corrections (BH-FDR) are appropriately applied.
Notebook Organization: All notebooks follow a clear structure (imports → data processing → analysis → visualization → save results). The progression from cohort assembly (NB01) → feature extraction (NB02) → baseline construction (NB03) → hypothesis tests (NB04-06) is logical and well-documented.
Data Pipeline: Excellent separation of Spark-dependent data generation from local analysis. Intermediate outputs (.parquet, .tsv) enable downstream notebooks to run independently. The pipeline correctly caches Spark DataFrames and uses .toPandas() only on final aggregated results.
Findings Assessment
Conclusions vs Evidence: All conclusions are well-supported by the data presented. The H3 finding (porewater bias) is particularly strong with overwhelming statistical evidence (p=4×10⁻¹²). The H2 partial support appropriately distinguishes between cohort-level and within-phylum effects. The H1 negative result is honestly reported with proper discussion of potential mechanisms.
Limitations Acknowledged: The project transparently acknowledges key limitations including small sample sizes, cultivation bias as both finding and confounder, compartment annotation uncertainty, and GapMind ceiling effects. Statistical power limitations are appropriately discussed.
Phylogenetic Control: The within-phylum decomposition for H2 is methodologically sophisticated and reveals that only sulfate reduction (not Wood-Ljungdahl or hydrogenase) survives phylogenetic control. This level of analytical rigor is exemplary.
Literature Integration: Findings are appropriately contextualized against the source literature. The direct replication of Bagnoud (2016) and divergence from Mitzscherling (2023) provides compelling validation of the cultivation bias hypothesis.
Suggestions
-
Emphasize sample size limitations earlier: While limitations are discussed in the REPORT.md, the README could include a brief note that statistical power is limited to detecting large effects (Cohen's d > 0.7) due to small cohort sizes.
-
Consider sensitivity analysis for compartment annotation: The keyword-based compartment classification could benefit from a sensitivity analysis using stricter definitions, particularly for the "porewater_borehole" vs "rock_attached" distinction that is central to H3.
-
Add genome size correlation analysis: Since genome size correlates with biosynthetic completeness, a brief correlation analysis in H1 could help distinguish biological vs methodological effects on the self-sufficiency metric.
-
Future work prioritization: Consider ranking the suggested future directions by feasibility/impact. The MAG-augmented expansion suggestion is excellent but could benefit from specific collection recommendations (e.g., which Mont Terri MAG datasets to prioritize).
-
Cross-reference with ENIGMA data: Given the Oak Ridge connection mentioned in the literature review, a brief discussion of how findings might relate to ENIGMA subsurface datasets in BERDL could strengthen the synthesis.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
H1 Self Sufficiency Violin
H2 Toolkit By Cohort
H3 Porewater Vs Rock
Notebooks
01_cohort_assembly.ipynb
01 Cohort Assembly
View notebook →
02_genome_features.ipynb
02 Genome Features
View notebook →
03_soil_baseline.ipynb
03 Soil Baseline
View notebook →
04_h1_self_sufficiency.ipynb
04 H1 Self Sufficiency
View notebook →
05_h2_anaerobic_toolkit.ipynb
05 H2 Anaerobic Toolkit
View notebook →
06_h3_porewater_bias.ipynb
06 H3 Porewater Bias
View notebook →